Single Step Free Energy Perturbation (SSFEP)¶
Background¶
Free energy perturbation (FEP) has long been considered the gold standard in calculating relative ligand-binding free energies. However, FEP is often impractical for evaluating large number of changes to a parent ligand due to the large computational cost. Single Step Free Energy Perturbation (SSFEP) is an alternative that can be orders of magnitude faster than conventional FEP when evaluating large number of changes to a parent ligand, while maintaining useful accuracy for small functional group modifications [6].
The SSFEP method involves post-processing of MD simulation data of a ligand in a given environment in the canonical ensemble to estimate the alchemical free energy change of chemically modifying the ligand. Zwanzig’s FEP formula is used,
where \(k_\mathrm{B}\) is the Boltzmann constant and \(T\) is the
temperature. The angular brackets indicate an average of the exponential factor
over the MD trajectory of ligand \(L1\) in the given environment, env,
which can be either the solvated protein or water. \(\Delta
E\) is the energy difference between the two systems involving L1 and L2, which
in practice is computed as the difference in the interaction energies of the
two ligands in the corresponding environment:
The environment env in each system is defined as all atoms with the
exception of ligand atoms. As the environment is constant between the two
ligands, the internal environmental energy cancels exactly during the
computation of \(\Delta E\). In addition, as the difference between L1 and
L2 involves a very small number of heavy atom modifications, we expect any
differential intra-ligand energy terms to also cancel exactly between the
solution and protein environments. Therefore, once \(\Delta
G_{L1\rightarrow L2}^\mathrm{protein}\) and \(\Delta G_{L1\rightarrow
L2}^\mathrm{water}\) are computed according to Eq. (1),
the relative binding free energy is given by
The SSFEP approach allows the data from simulation of a single protein-ligand complex to be rapidly post-processed to evaluate tens to hundreds of potential modifications involving multiple sites on the parent ligand. Given this, the best results are achieved when SSFEP is used to evaluate small modifications to the parent ligand.
In a recent study [7], the ability of standard FEP and SSFEP to reproduce the experimental relative binding affinities of known ligands for two proteins, ACK1 and p38 MAP kinase, was tested. SSFEP was able to produce comparable results to ful FEP while requiring a small fraction of the computational resources.
Usage¶
The following usage details are provided for completeness. We strongly recommend using the SilcsBio GUI to set up, run, and analyze SSFEP calculations as described in quickstart_gui/quickstart_gui.
Setup SSFEP Simulation¶
To perform the SSFEP precomputation simulations, protein coordinates in PDB file format and parent ligand coordinates in mol2 file format are required. The protein should have termini properly capped, missing loops built or the ends of the missing loops capped, standard atom and residue names, and sequential atom and residue numbering. Using these two files, run the following:
${SILCSBIODIR}/ssfep/1_setup_ssfep prot=<Protein PDB> lig=<Ligand Mol2/SDF>
Warning
The setup program internally use the GROMACS utility pdb2gmx,
which may have problems processing the protein PDB file. The most
common pdb2gmx issue involves mismatches between the
expected residue name/atom names in the input PDB and those
defined in the CHARMM force-field.
To fix this problem: Run the pdb2gmx command manually from
within the 1_setup directory for a detailed error message.
Please contact support@silcsbio.com for additional assistance.
Following completion of the setup, run 10 MD jobs:
${SILCSBIODIR}/ssfep/2_run_md_ssfep prot=<Protein PDB> lig=<Ligand Mol2/SDF>
This command will submit 10 jobs to the pre-defined queue: 5 for the ligand in water and 5 for the ligand complexed with protein.
Once the precomputation simulations are completed, the
2_run_md/1_lig/[1-5] and 2_run_md/2_prot_lig/[1-5]
directories will contain *.1-10.whole.trr trajectory files. If
these files are not generated, then your simulations are either still
running or have stopped due to a problem. Look into the log files
within these directories for an explanation of the failure.
Prepare chemical group transformations¶
Create a text file modifications.txt with instructions
listing the desired modifications to the parent ligand.
Three types of modifications can be performed, as listed in the table.
| Command | Modification |
|---|---|
| JOIN | Join a fragment (mol2) to the parent at a defined site |
| REPL | Replace an atom at a defined site on the parent with a fragment (mol2) |
| MUDE | Change an atom at a defined site on the parent to another atom-type |
An example ligand modification input file is provided in examples/ssfep/modification.txt file. For help on how to define these operations, run the
following command:
${SILCSBIODIR}/programs/chemmod -h
Access the groups directory from the following location to look at all the possible fragments that can be added/joined to the parent ligand:
${SILCSBIODIR}/data/groups
Modifications JOIN example¶
A JOIN operation can be performed between the parent and a ch4.mol2
fragment by adding the following line to modifications.txt:
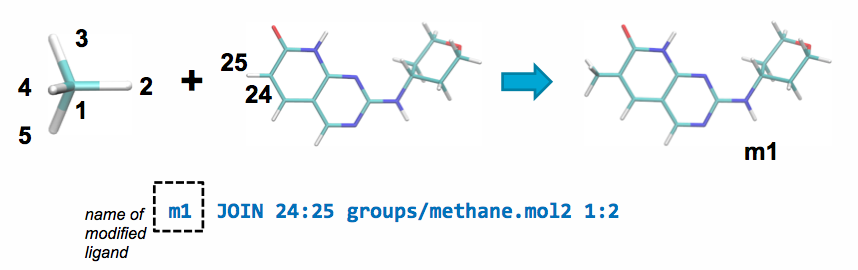
This line will join atom 24 in the parent ligand with
atom 1 in methane.mol2 and delete atoms 25 and 2 in the parent ligand and
the joined fragment, respectively.
Modifications REPL example¶
A REPL operation can be performed between the parent and a ch4.mol2
fragment by adding the following line to modifications.txt:
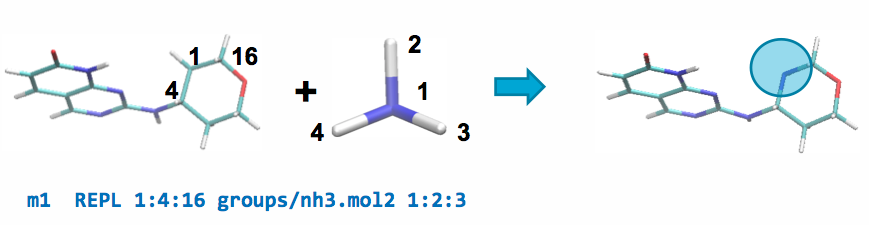
This line will replace atom 1 of the parent ligand with atom 1 of the fragment by aligning atoms 2 & 3 of the fragment with atoms 4 & 16 of the parent, respectively, and replacing the carbon in the ring with a nitrogen atom.
Modifications MUDE example¶
The same transformation in the previous section can also be achieved using a MUDE operation:
m2 MUDE MU 1:7 DE 21
This line will mutate atom 1 (atom index number) in the parent with nitrogen (atom index number 7) along with deleting the hydrogen (atom index number 21) attached to the parent carbon.
Ligand decoration¶
To evaluate multiple modifications to a single site on the parent ligand, use the following syntax:
m1 JOIN 24:25 ssfep_lig_decoration_master_modification.txt
Copy ssfep_lig_decoration_master_modification.txt from the
${SILCSBIODIR}/data/ folder. This master modification list contains
70 commonly-used modifications. Be careful to pay attention to
chemistry; if some of these modifications are not suitable for a site, you can
comment them out using ! at the beginning of that line.
This single entry in your modifications.txt will generate 70
separate modifications to the parent ligand, each with a prefix m1_
Evaluating binding affinity changes¶
Once modifications.txt has been prepared and the MD simulations
involving the parent ligand are completed, run the following script
to set up a \(\Delta \Delta G\) calculation.
${SILCSBIODIR}/ssfep/3a_setup_modifications prot=<Protein PDB> lig=<Ligand Mol2/SDF File> mod=modifications.txt
This will submit 10 jobs to evaluate all snapshots from the completed MD
simulations of the parent ligand in order to calculate the
change in free energy for every modification specified in your
modifications.txt. Structures of these modifications in mol2 format
are output as 3_analysis_<modified ligand name entry in
modifications.txt>/mod_files/*.mol2.
After these jobs complete, you may obtain \(\Delta \Delta G\) for your full list of modifications using:
${SILCSBIODIR}/ssfep/3b_calc_ddG_ssfep mod=modifications.txt
Example output follows:
