SILCS-PPI
Protein-Protein Interactions (PPI) energy function based on the Site-Identification by Ligand Competitive Saturation (SILCS) framework and utilize the fast Fourier transform (FFT) correlation approach. The free energy content of the SILCS FragMaps represent an alternative to traditional energy grids and they can be efficiently utilized to guide FFT-based protein docking.
SILCS-PPI Using the SilcsBio GUI
Coming soon!
SILCS-PPI Using the CLI
The SILCS-PPI workflow can be run with the following steps:
Set up and run PPI calculations:
User must have completed the SILCS simulations and generated FragMaps for the target proteins. If self interactions are to be considered, the same protein must be specified for both
prot1andprot2. Typically, the first protein is the “receptor” and the second protein is the “ligand”.$SILCSBIODIR/ppi/1_setup_ppi prot1=<first prot pdb> mapsdir1=<location and name of directory containing FragMaps> prot2=<second prot pdb> mapsdir2=<location and name of directory containing FragMaps>
Required parameters:
Path and name of first protein PDB file:
prot1=<first protein PDB>
Path and name of directory containing FragMaps for the first protein:
mapsdir1=<location and name of directory containing FragMaps>
Path and name of second protein PDB file:
prot2=<second protein PDB>
Path and name of directory containing FragMaps for the second protein:
mapsdir2=<location and name of directory containing FragMaps>
Optional parameters:
Path and name of directory for PPI results:
ppidir=<folder where PPI result is stored; default: 3_ppi>
Rotation step size for sampling second protein orientation in reference to the first protein:
rotsize=<rotation step size; 10/15/20; default=10>
A pre-defined set of relative transformations based on the
rotsizeparameter are selected from the$SILCSBIODIR/data/ppi_rot_anglesdirectory.Number of solutions per rotation:
numsol=<number of solutions per rotation; default=10>
Bundle multiple (single) jobs into larger jobs:
bundle=<bundle multiple (single) jobs into larger jobs; true/false; default=true>
If
bundle=true, the number of jobs is determined by thenjobsparameter. Ifbundle=false, each job will be submitted separately.Total number of jobs used when
bundle=true:njobs=<total # of jobs used when bundle=true; default=16>
Number of processors per job:
nproc=<number of processors per job; default=8>
Restart a job for specific bundle:
restart=<restart a job for specific bundle; e.g. restart=#1; default=false>
If
restart=#<bundle_number>, the job will be restarted for the specified bundle number.
Additional parameters:
Using Full Size of fragmap for PPI calculations:
fullmap=<flag for using full map or not; true/false; default=true>
If
fullmap=false, the center and radius parameters must be set.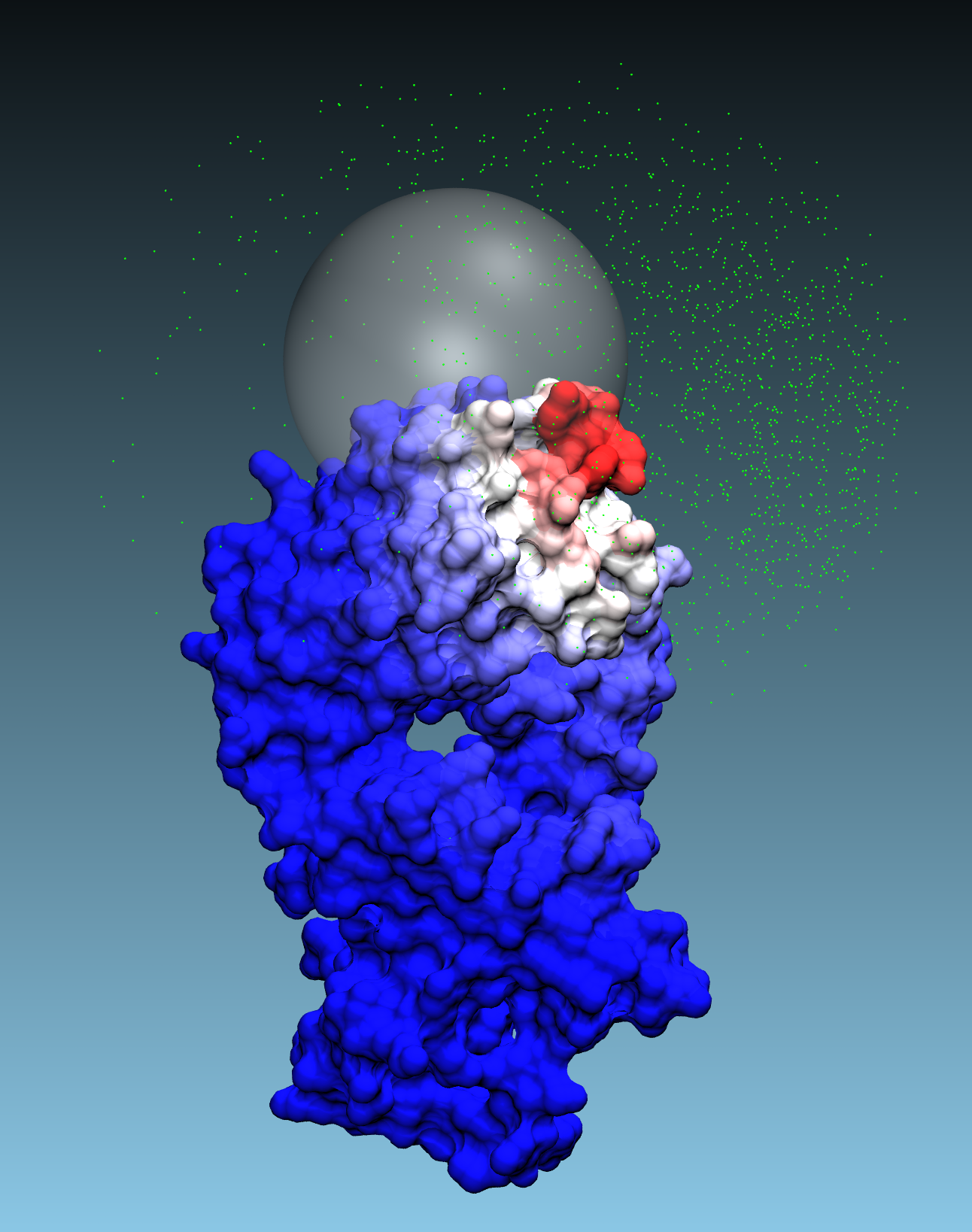
Center for transformation parameter:
center="x,y,z"; e.g. center="20,30,-2"
The center is the point on the surface of the first protein around which a sphere of radius
radiuswill be defined. Only the poses of the second protein that are interfacing with at least one of residues of the first protein within the specified radius will be considered for PPI calculations. Iffullmap=false, this parameter must be set.Radius for transformation parameter:
radius=<X Angstroms>
The solutions will be restricted to the points on the surface of the first protein that are within the specified radius from the center. If
fullmap=false, this parameter must be set.Truncate maps and remove overlaps:
filter=<truncate maps and remove overlaps; true/false; default=true>
If
filter=true, the maps will be truncated by setting the GFE values to zero for all points that are not within the cutoff distance from the surface of the exclusion map. Iffilter=false, the maps will not be truncated and all points will be used in the PPI calculations which may lead to longer runtimes and larger output files.Cutoff for filtering:
cutoff=<cutoff for filtering; default=5>
The cutoff distance is used to determine which points are within the range from the surface of the exclusion map. If
filter=true, the GFE values for all points that are not within the cutoff distance from the surface of the exclusion map will be set to zero.Custom parameter file:
paramsfile=<custom parameter file; default: $SILCSBIODIR/templates/ppi/params.tmpl>
The custom parameter file can be used to override the default parameters used for PPI calculations. If not specified, the default parameter file will be used.
Path to Python executable:
python=<path to python executable for filtering maps; default=$(which python)>
The path to the Python executable used for filtering maps. If not specified, the default Python executable identified by using the
which pythoncommand will be used.
Collect PPI results and perfor clustering:
Once the PPI calculations are complete, the results can be collected and clustered to identify the most favorable binding poses of the second protein relative to the first protein.
$SILCSBIODIR/ppi/2_collect_ppi prot1=<first prot pdb> prot2=<second prot pdb>
Required parameters:
Path and name of first protein PDB file:
prot1=<first protein PDB>
Path and name of second protein PDB file:
prot2=<second protein PDB>
Optional parameters:
Path and name of directory where PPI results are stored:
ppidir=<folder where PPI result is stored; default: 3_ppi>
Perform clustering:
cluster=<perform clustering; true/false; default=true>
If
cluster=true, the PPI results will be clustered to identify the most favorable binding poses of the second protein relative to the first protein.Number of top clusters to output:
topn=<number of top clusters to output; default=2000>
Path to Python executable for clustering:
python=<path to python executable for clustering; default=$(which python)>
Re-build PPI complex PDB files:
After the PPI results have been collected and clustered, the final step is to rebuild the PPI complex PDB files for visualization and further analysis.
$SILCSBIODIR/ppi/3_build_cmpx_pdb prot1=<first prot pdb> prot2=<second prot pdb>
Required parameters:
Path and name of first protein PDB file:
prot1=<first protein PDB>
Path and name of second protein PDB file:
prot2=<second protein PDB>
Optional parameters:
Path and name of logfile from the previous step (2_collect_ppi):
logfile=<ppi logfile from 2_collect_ppi; default=ppips_clusters.log>
Prefix for output PDB files:
prefix=<prefix for output pdb files; default=complex>