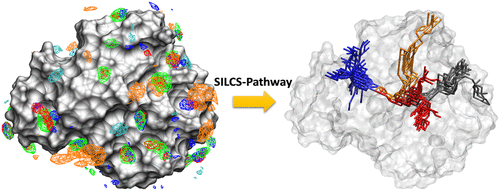
SILCS-Pathway
Background
SILCS-Pathway is a computational workflow designed to identify and analyze ligand dissociation pathways from protein or RNA binding sites. Unlike traditional approaches that focus solely on binding affinity, SILCS-Pathway leverages the correlation between drug efficacy and dissociation kinetics. The method builds on the Site Identification by Ligand Competitive Saturation (SILCS) technique, which precomputes FragMaps—free energy landscapes for various chemical functionalities around a target biomolecule.
SILCS-Pathway utilizes the A* pathfinding algorithm to enumerate all possible ligand dissociation routes from the binding site to the bulk solvent. The search is performed on a set of evenly distributed points around the protein, generated using a Fibonacci lattice. The cost function for the A* algorithm incorporates SILCS exclusion maps and grid free energy scores, enabling the identification of pathways that reflect local protein flexibility and favorable ligand interactions.
By systematically traversing all bulk solvent points, SILCS-Pathway locates and clusters all feasible dissociation pathways, providing a comprehensive view of ligand unbinding mechanisms. The approach has been validated against proteins previously studied with enhanced sampling molecular dynamics, demonstrating its ability to efficiently capture key dissociation routes. The identified pathways serve as a foundation for subsequent determination of ligand dissociation kinetics using SILCS free energy profiles. Additional details on SILCS-Pathway are available in reference [23].
SILCS-Pathway Using the SilcsBio CLI
There are six main steps in the SILCS-Pathway workflow, each of which can be executed using the SilcsBio CLI. Below is a detailed description of each step, including usage instructions, required and optional parameters, expected outputs, and important notes for successful execution.
To run all six steps of the SILCS-Pathway workflow sequentially, use the following command:
${SILCSBIODIR}/silcs-pathway/run_silcs_pathway prot=<protein PDB file> center="x,y,z"
This script automates the complete SILCS-Pathway process, executing each step in order and handling intermediate files as needed. It is recommended for users who want a streamlined, end-to-end workflow.
Required parameters
prot: Full path to the protein PDB file.center: Coordinates (x,y,z) of the binding site center (e.g., center=”20.1,33,-2”).
Optional parameters
mapsdir: Directory containing FragMaps (default: maps).outputdir: Output directory for results (default: 4_pathway).batch: Submit jobs to queuing system (default: true).
Alternatively, you may execute each step individually as described below:
Fibonacci Sphere Generation
This step generates a defined number of Fibonacci spheres around a protein structure using the fibo program. The spheres are used in subsequent pathway analysis steps to sample spatial positions around the protein.
Usage
${SILCSBIODIR}/silcs-pathway/1_cal_fibo prot=<protein PDB file> [numfibo=<number>] [outputdir=<directory>] [cleanup=<true/false>] [batch=<true/false>]Required parameters
prot: Full path to the protein PDB file.Optional parameters
numfibo: Number of Fibonacci spheres to generate (default: 500).
outputdir: Output directory for results (default: 4_pathway).
cleanup: Whether to clean up intermediate files (default: true).
batch: Submit job to queuing system (default: false).Output
Fibonacci sphere coordinates and log file in the specified output directory.
If successful, a message confirming completion is displayed.
If incomplete, a warning is shown with the number of spheres generated.
Notes
The script checks for valid input files and parameters.
The script checks for valid input files and parameters.
If the output directory exists, you can choose to overwrite, backup, or cancel.
Errors and warnings are printed for missing files, invalid parameters, or failed sphere generation.
A* Search Pathway Analysis
This step performs pathway analysis using the A* search algorithm to find optimal pathways for ligand binding or unbinding. It utilizes the generated Fibonacci spheres and the protein structure, starting from a defined binding site center.
Usage
${SILCSBIODIR}/silcs-pathway/2_astar prot=<protein PDB file> center="x,y,z" [outputdir=<directory>] [mapsdir=<FragMaps directory>] [cleanup=<true/false>] [batch=<true/false>]Required parameters
prot: Path to the protein PDB file.
center: Coordinates (x,y,z) of the binding site center (e.g., center=”20.1,33,-2”).Optional parameters
outputdir: Output directory for results (default: 4_pathway).
mapsdir: Directory containing FragMaps (default: maps).
cleanup: Whether to clean up intermediate files (default: true).
batch: Submit job to queuing system (default: false).Output
Pathway analysis results, including pathway coordinates and PDB files, in the specified output directory.
Log files for each search and summary of successful/failed points.
If successful, a message confirming completion is displayed.
If incomplete, warnings and logs are provided for debugging.
Notes
The script checks for valid input files, parameters, and required directories.
The output directory must contain results from step 1 (fibo_data.txt).
Errors and warnings are printed for missing files, invalid parameters, or failed A* search.
Failed pathway points are handled and logged for further analysis.
Truncated Pathway Analysis
This step processes the pathway points generated from the A* search and removes those that are outside the protein structure, resulting in a truncated pathway that is physically relevant for further analysis.
${SILCSBIODIR}/silcs-pathway/3_truncate_path prot=<protein PDB file> [outputdir=<directory>] [batch=<true/false>] [cleanup=<true/false>]Required parameters
prot: Path to the protein PDB file.Optional parameters
outputdir: Output directory for results (default: 4_pathway).
batch: Submit job to queuing system (default: false).
cleanup: Whether to clean up intermediate files (default: true).Output
Truncated pathway coordinates and PDB files in the specified output directory (coors/ and pathpdbs/).
Log file indicating success or any issues encountered (truncate_<protein>.log).
If successful, a message confirming completion is displayed.
If incomplete, warnings and logs are provided for debugging.
Notes
The script checks for valid input files, parameters, and required directories.
The output directory must contain successful results from step 2 (astar_<protein>.log with “success”).
Errors and warnings are printed for missing files, invalid parameters, or failed truncation.
Pathway points outside the protein are removed to ensure physical relevance for downstream analysis.
Distance Matrix Calculation
This step calculates the distance matrix between all truncated pathway points, providing a quantitative basis for further pathway analysis and clustering.
Usage
${SILCSBIODIR}/silcs-pathway/4_cal_dmatrix prot=<protein PDB file> [outputdir=<directory>] [batch=<true/false>] [cleanup=<true/false>]Required parameters
prot: Path to the protein PDB file.Optional parameters
outputdir: Output directory for results (default: 4_pathway).
batch: Submit job to queuing system (default: false).
cleanup: Whether to clean up intermediate files (default: true).Output
Distance matrix file (distmat.dat) in the specified output directory.
Log file indicating success or any issues encountered (dmatrix_<protein>.log).
If successful, a message confirming completion is displayed.
If incomplete, warnings and logs are provided for debugging.
Notes
The script checks for valid input files, parameters, and required directories.
The output directory must contain successful results from step 3 (truncate_<protein>.log with “success”).
Errors and warnings are printed for missing files, invalid parameters, or failed distance matrix calculation.
The distance matrix is used for downstream pathway clustering and analysis.
Clustering Pathway Points
This step clusters the truncated pathway points using the cluster program and removes duplicate PDBs, resulting in a set of unique pathway conformations for further analysis.
Usage
${SILCSBIODIR}/silcs-pathway/5_cluster prot=<protein PDB file> [cutoff=<cutoff value>] [outputdir=<directory>] [batch=<true/false>] [cleanup=<true/false>]Required parameters
prot: Path to the protein PDB file.Optional parameters
cutoff: Clustering cutoff value in Angstroms (default: 20.0).
outputdir: Output directory for results (default: 4_pathway).
batch: Submit job to queuing system (default: false).
cleanup: Whether to clean up intermediate files (default: true).Output
Clustered and unique pathway PDB files in the specified output directory (pathunipdbs/).
Clustering results file (cl.result) and log file (cluster_<protein>.log).
If successful, a message confirming completion is displayed.
If incomplete, warnings and logs are provided for debugging.
Notes
The script checks for valid input files, parameters, and required directories.
The output directory must contain successful results from step 4 (dmatrix_<protein>.log with “success” and distmat.dat).
Errors and warnings are printed for missing files, invalid parameters, or failed clustering.
Duplicate PDBs are removed to ensure a unique set of pathway conformations for downstream analysis.
Visualization Script Generation
This step generates VMD and PyMOL scripts to visualize the clustered pathway conformations, enabling graphical inspection and presentation of the results.
Usage
${SILCSBIODIR}/silcs-pathway/6_gen_visuals prot=<protein PDB file> [clsize=<cluster size cutoff>] [outputdir=<directory>] [batch=<true/false>] [cleanup=<true/false>]Required parameters
prot: Path to the protein PDB file.Optional parameters
clsize: Minimum number of members in a cluster to be included in the visualization (default: 2).
outputdir: Output directory for results (default: 4_pathway).
batch: Submit job to queuing system (default: false).
cleanup: Whether to clean up intermediate files (default: true).Output
VMD script (pathcluster-cutoff-<clsize>.vmd) and PyMOL script (pathcluster-cutoff-<clsize>.pml) in the specified output directory.
Log messages indicating successful script generation.
If successful, a message confirming completion is displayed.
If incomplete, warnings and logs are provided for debugging.
Notes
The script checks for valid input files, parameters, and required directories.
The output directory must contain successful results from step 5 (cluster_<protein>.log with “success”).
Errors and warnings are printed for missing files, invalid parameters, or failed script generation.
Both VMD and PyMOL scripts are generated for flexible visualization options.
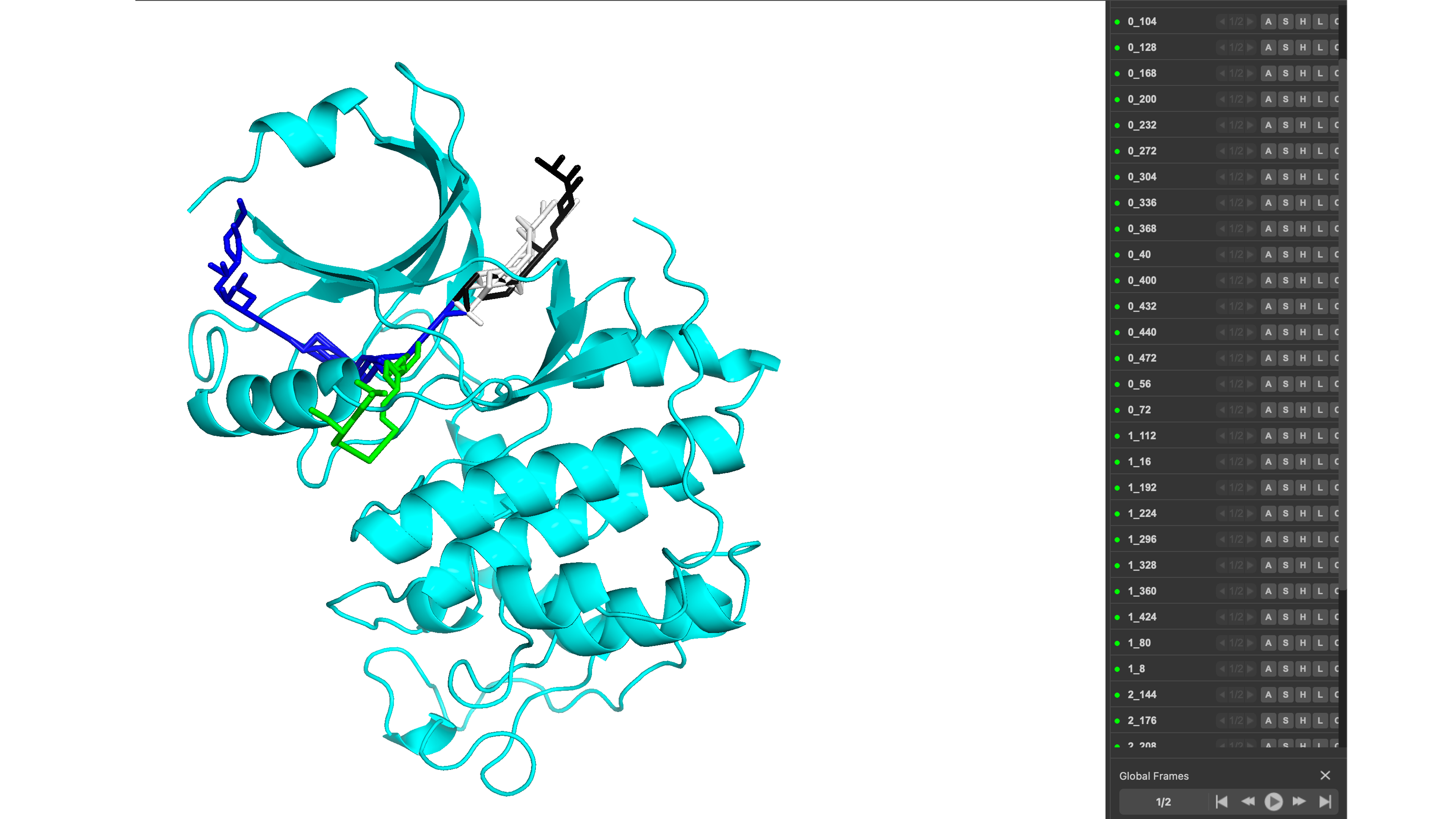
The script names the clusters based on the specific cluster and pathway name with clusterid_pathwayid.pdb for easy identification.
An example of the visualization in pymol can be seen below: