SILCS-MC: Docking and Pose Refinement¶
Background¶
The power of SILCS lies in the ability to use FragMaps to rapidly evaluate binding of diverse ligands to a target. SILCS-MC is Monte-Carlo (MC) sampling of ligands in translational, rotational, and torsional space in the field of FragMaps. MC sampling uses CGenFF intramolecular energies and the Ligand Grid Free Energy (LGFE), which is the sum of atomic Grid Free Energies (GFEs). The Exclusion Map prevents ligand sampling where no probe or water molecules visited during SILCS simulations. SILCS-MC allows for rapid conformational sampling of the ligand while accounting for protein flexibility in a mean-field-like fashion since ligand affinity and volume exclusion are embedded in the combination of FragMaps and the Exclusion Map. Final ligand scoring is based on the LGFE score [12][14].
SILCS-MC can be used to generate and score binding poses for a ligand to a target using SILCS FragMaps that have been previously computed for that target (see SILCS: Site Identification by Ligand Competitive Saturation). This can readily be done for a single ligand or a database of ligands. Two default conformational sampling protocols are available, “docking” and “pose refinement,” as described in detail below. The docking protocol is useful when no information is available about the binding pose, as it entails extensive translational, rotational, and intramolecular conformational sampling. The pose refinement protocol is useful when a reasonable starting pose for the ligand is available, for example to re-score poses output by another high-throughput in silico screening tool. Instructions for developing custom SILCS-MC ligand conformational sampling protocols are provided at the end of this chapter.
Running SILCS-MC docking¶
Docking is exhaustive sampling of a ligand’s conformation in a given pocket to determine its most favorable orientation and internal geometry as defined by LGFE scoring. The pocket is predefined as a 10 Å sphere and the center of the pocket is taken as either the center of the supplied ligand molecule coordinates or explicitly given by the user. This protocol entails five independent MC runs with the ligand.
This protocol is recommended for ligands with diverse chemotypes and unknown binding poses. When the pose of a parent ligand is known and SILCS-MC evaluations are to be performed over a congeneric series, the pose refinement protocol is recommended instead (see below).
Running SILCS-MC docking from the SilcsBio GUI¶
Select New SILCS-MC Docking project from the Home page.
Enter a project name and select your remote server. On the remote server, select the “Project Location” directory. Output files will be placed in a subdirectory
3_silcsmc/in the Project Direcory. Select the “FragMap Location,” which is where the FragMaps from the prior SILCS job are located on the server. Likewise, select the “Protein PDB File,” which was the input PDB file for the SILCS jobs. Finally, select the “Ligand SDF File.” This is the file on your local computer that contains the database of ligands to be docked.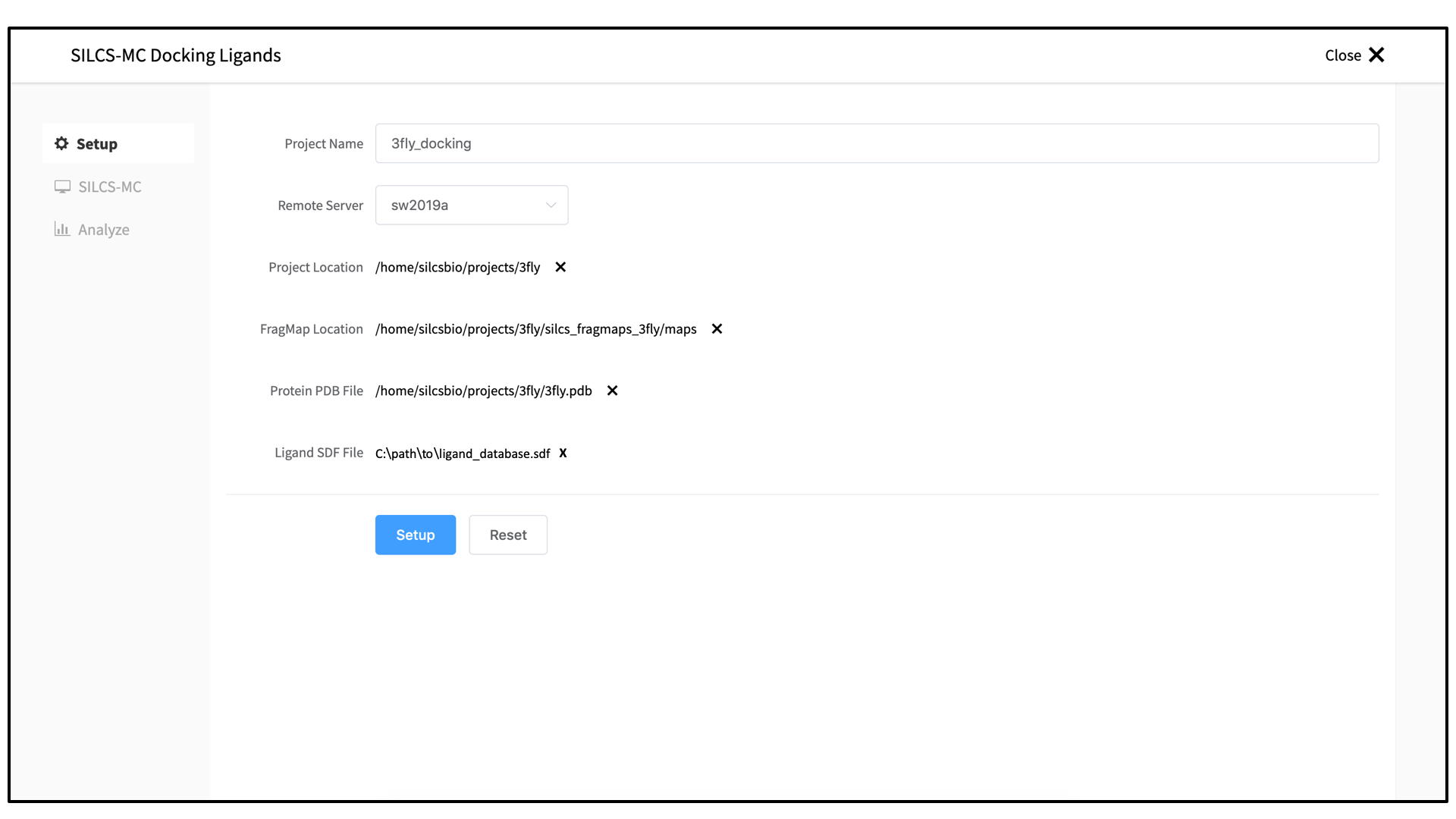
Once all information is entered correctly, press the “Setup” button at the bottom of the page. The page will update to list the number of ligands and show options for the sampling protocol (“Docking” or “Pose Refinement”) and the sampling region. Select the “Docking” option and then press the “Select Pocket” button.
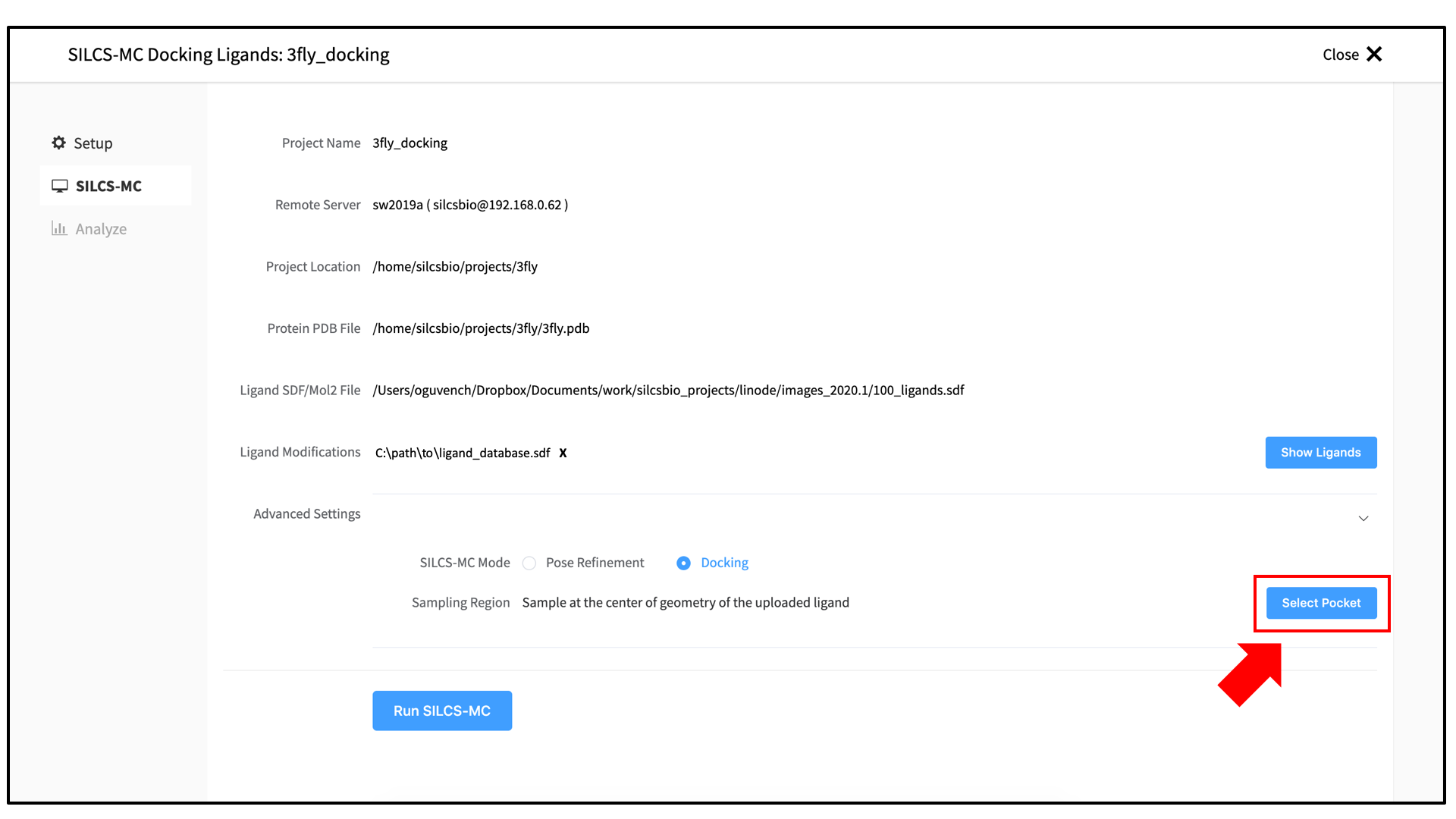
The GUI will now be showing the protein molecular graphic in the center pane. On the right-hand side, select the “Pocket” tab. From this tab, you can define the pocket center based on the center-of-geometry of a ligand pose (“Define Pocket using Ligand”), or a target residue selection (“Define Pocket using Selection”), or by directly entering an x, y, z coordinate (“Define Pocket by XYZ”). You will also need to choose a radius (default value “10”) to complete the definition of the spherical pocket. If it is difficult to see the spherical pocket definition in the center pane, click on the FragMap tab on the right-hand side and deselect the “Protein Surface” to hide the protein surface.
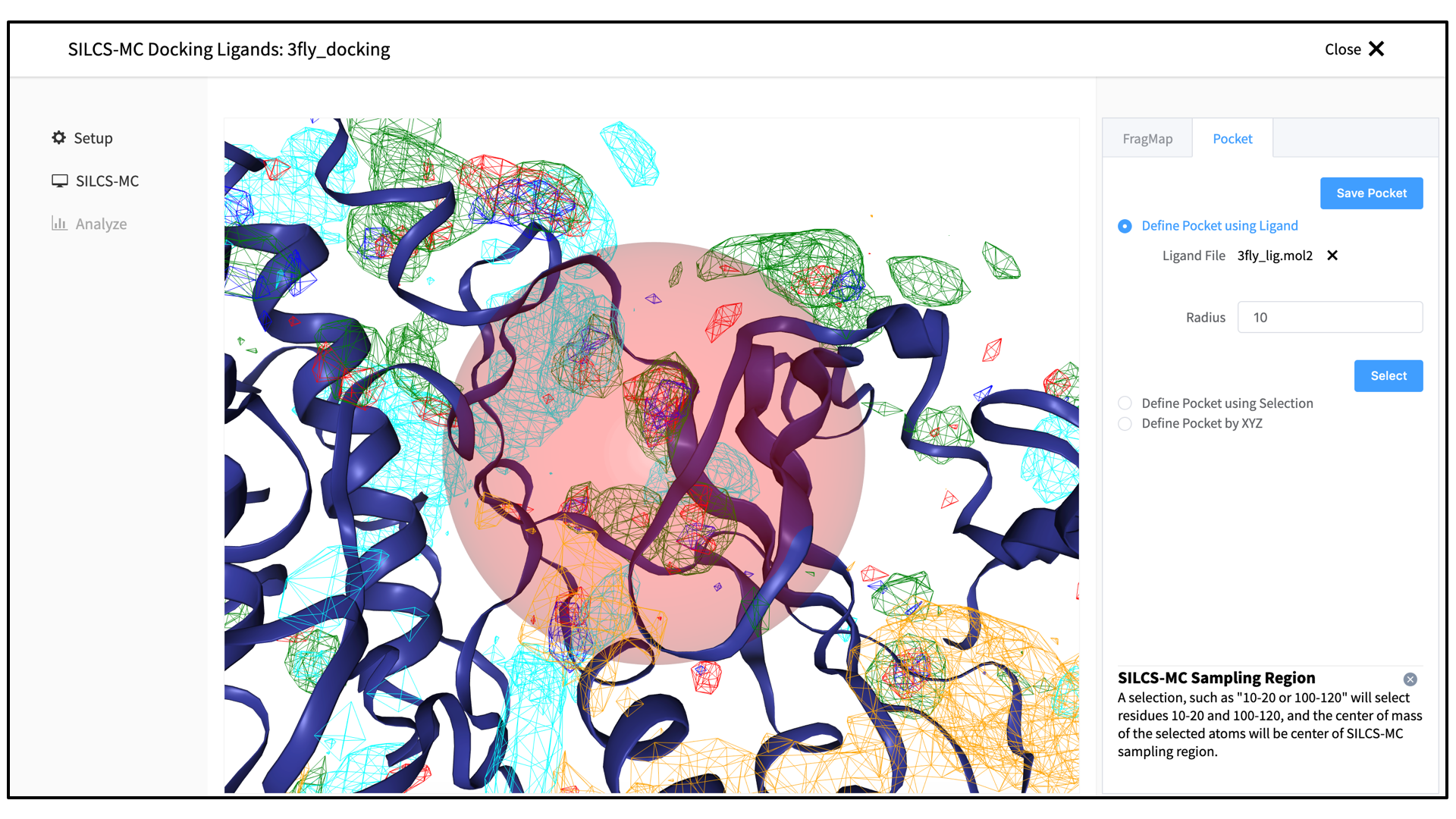
Click on the “Save Pocket” button to continue, and you will be returned to the previous screen, which now includes “Sampling Region” information consisting of the spherical pocket center and radius. You may now click the “Run SILCS-MC” button to start the SILCS-MC docking. Doing so will submit jobs to the remote server job schedular and list them in a pop-over window.
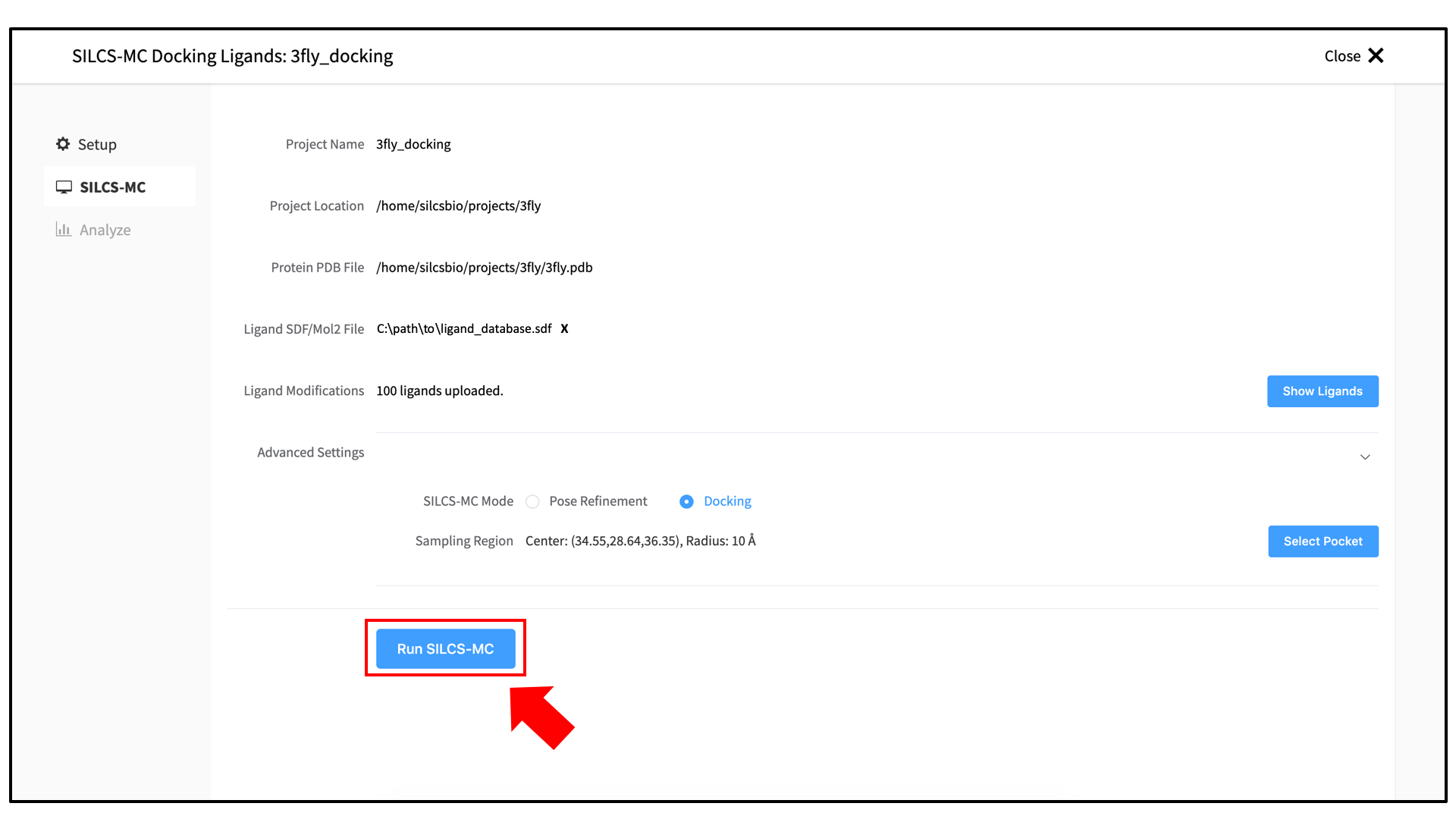
Once all jobs are submitted, you may click on the “Setup Successful” button to dismiss the pop-over window and return to the previous screen, which will now show a “Simulation Progress” section. You may update this section by scrolling to the bottom of the screen and clicking the “Refresh” button. This will update the progress bars for all of the ligands being docked.
Once progress bars for all ligands reach 100%, click on the “Show Chart” button above the “Simulation Progress” section to proceed.
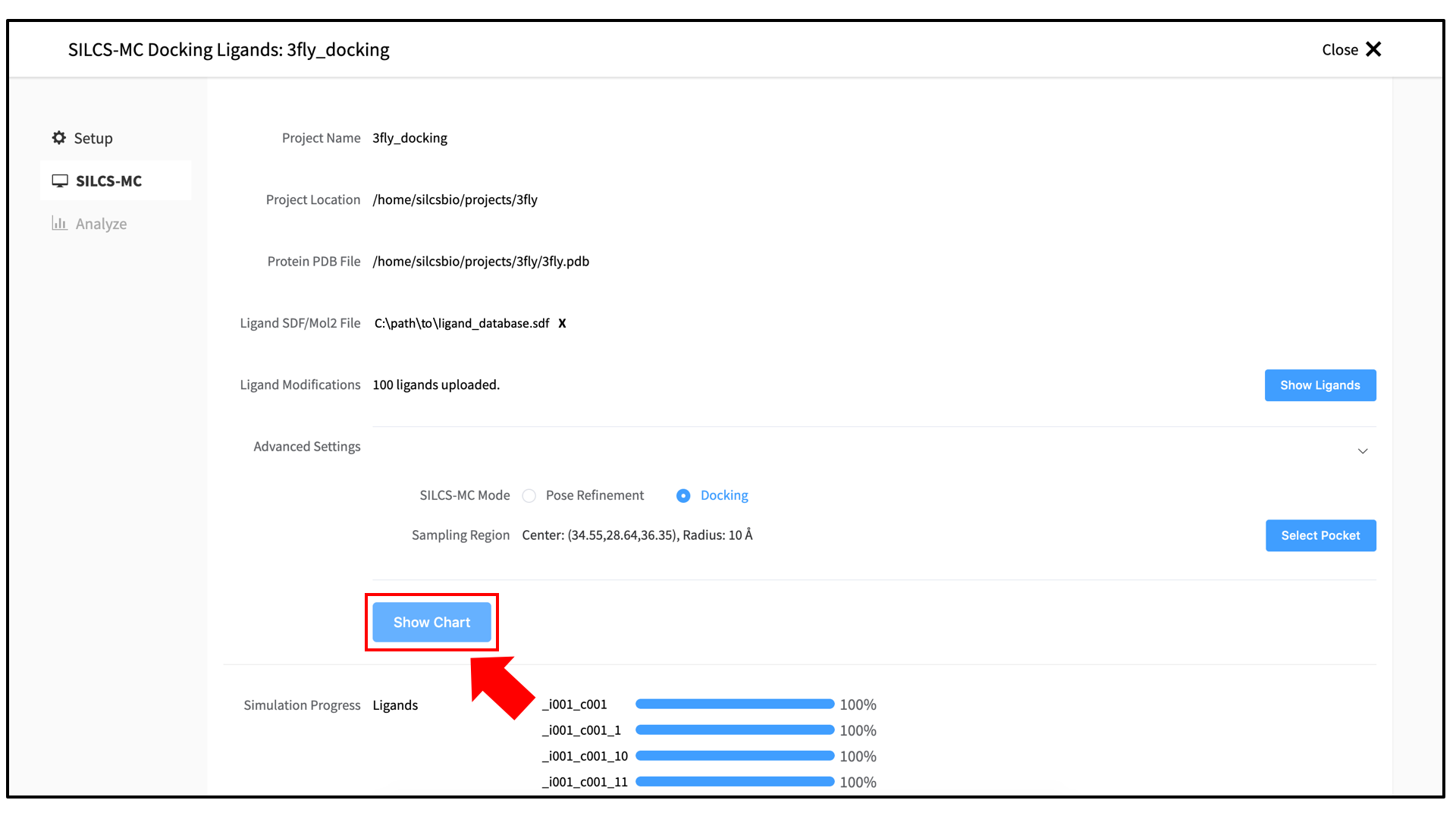
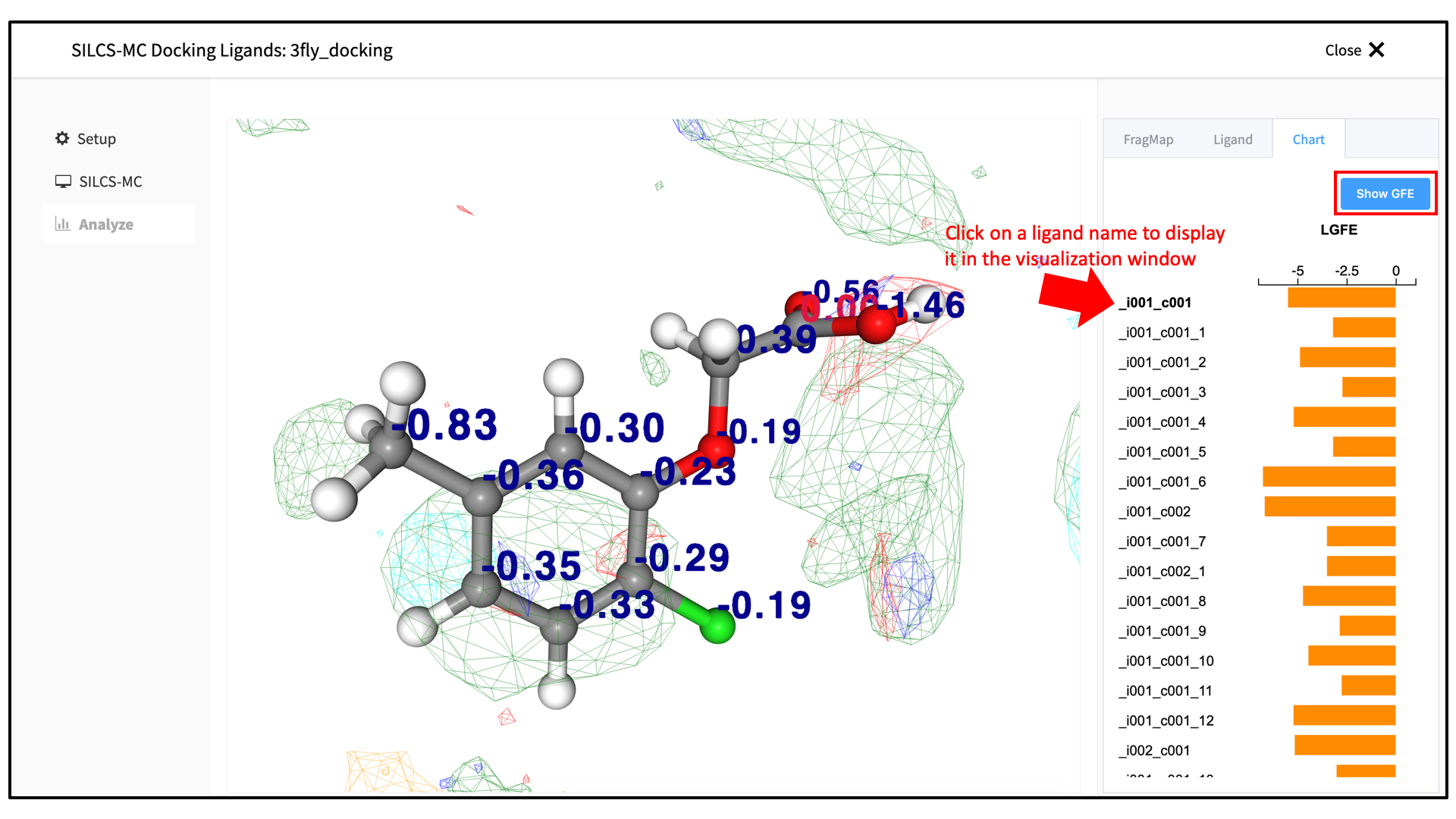
Upon successful completion of this command in the pop-over window, you may click on the green “Data collection finished” button to return to the GUI. A new tab, labeled “Chart” will have been created in the right-hand panel. Under that tab, clicking on a ligand name will center the visualization window at the ligand and also display the protein surface and FragMap densities. For ease of viewing of the molecular graphics, you may wish to deselect the “Protein Surface” and “Protein Cartoon” representations under the “FragMap” tab in the right-hand panel. After returing to the “Chart” tab and clicking on a ligand name, click on the “Show GFE” button in the upper right-hand corner to display the atom-wise Grid Free Energy (GFE) contributions to the total Ligand Grid Free Energy (LGFE) shown in bar-graph format, also under the “Chart” tab. The GFEs will all be shown in the molecular visualization window as scalar numbers adjacent to their respective atoms.
Running SILCS-MC docking from the command line interface¶
To set up and run SILCS-MC docking from the command line interface, create a directory containing all the ligands to be evaluated. Each ligand can be stored as a separate SDF or Mol2 file. Alternatively, all the ligands can be combined into a single SDF file.
${SILCSBIODIR}/silcs-mc/1_run_silcsmc_exhaustive \
prot=<prot pdb> \
ligdir=<directory containing ligand mol2/sdf> \
mapsdir=<directory containing SILCS FragMaps> \
center="x,y,z"
Note
.sdf, .sd, or .mol2 files can be placed in the ligdir
directory, and SILCS-MC will read a single molecule from each file.
Note that if a file contains multiple molecules, use of the ligdir
option will result in only the first molecule in the file being processed.
If you have an SDF file with multiple molecules in it, replace
ligdir=<directory containing ligand mol2/sdf> with
sdfile=<path to sdfile> to process all molecules in the file.
SILCS-MC docking protocol details¶
Docking spawns five independent single-core serial jobs per ligand.
Each run involves a maximum of 250 cycles and a minimum of 50 cycles of
Monte Carlo/Simulated Annealing (MC/SA) sampling of the
ligand within the defined 10 Å sphere. Each of these 250 cycles consists of
10,000 steps of MC at a high temperature followed by 40,000 steps of
SA towards a lower temperature. At the beginning of
each cycle, the ligand will be reoriented within the predefined sphere. When
no sphere center is defined by the user, the ligand orientation in the input
SDF or Mol2 file will be used as the starting pose for each cycle.
The MC sampling has three types of moves: i) molecular translations with a
maximum step size of 1 Å, ii) molecular rotation with a maximum step size of
180 degrees, and iii) intramolecular dihedral rotations with a maximum
step size of 180 degrees. For intramolecular dihedral rotations, only the
rotatable dihedral angles are selected for MC moves. The lowest LGFE scoring
pose from the MC sampling is used as starting pose in the following SA
sampling. The SA sampling also involves the same three types of moves, but
with a smaller step size compared to the MC sampling: i) molecular
translations with a maximum step size of 0.2 Å, ii) molecular rotation with a
maximum step size of 9 degrees and, iii) intramolecular dihedral rotations with
a maximum step size of 9 degrees. The lowest LGFE scoring pose from the SA is
saved in a multi-frame PDB file: 3_silcsmc/<run>/pdb/<lig>_mc.<run>.pdb.
In each run, after a minimum of 50 MC/SA cycles, if the LGFE score difference between the top three poses (defined by lowest LGFE scores) are less than 0.5 kcal/mol, then that run is considered converged and terminated. If the top three scored poses are separated by more 0.5 than kcal/mol, the MC/SA procedure continues either until the convergence criterion is met or until a maximum of 250 MC/SA cycles have been completed.
Running SILCS-MC pose refinement¶
The pose refinement protocol is designed to limit conformational sampling near the ligand input pose supplied by the user. Pose refinement is appropriate when there is high confidence in the input parent ligand pose and SILCS-MC evaluations are to be performed over a congeneric series. The sphere center for the pocket definition is the center-of-geometry from the input ligand pose.
Running SILCS-MC pose refinement from the SilcsBio GUI¶
Please see the previous section on running SILCS-MC docking from the SilcsBio
GUI, and, in Step 3, select the “Pose Refinement” option. Note that there
will be no “Select Pocket” step, as pose refinement assigns the pocket center
based on the center-of-geometry of the input ligand. For multiple input
ligands in a single .sdf file, each ligand will have its own
center-of-geometry used to define the pocket center.
Running SILCS-MC pose refinement from the command line interface¶
To set up and run SILCS-MC pose refinement, create a directory containing all
the ligands to be evaluated. Each ligand can be stored as a separate .mol2
or .sdf file. Alternatively, all the ligands can be combined into a single
.sdf file.
${SILCSBIODIR}/silcs-mc/1_run_silcsmc_local prot=<prot pdb> \
ligdir=<directory containing ligand mol2/sdf> \
mapsdir=<directory containing SILCS FragMaps>
Note
.sdf, .sd, or .mol2 files can be placed in the ligdir
directory, and SILCS-MC will read a single molecule from each file.
Note that if a file contains multiple molecules, use of the ligdir
option will result in only the first molecule in the file being processed.
If you have an SDF file with multiple molecules in it, replace
ligdir=<directory containing ligand mol2/sdf> with
sdfile=<path to sdfile> to process all molecules in the file.
SILCS-MC pose refinement protocol details¶
Pose refinement spawns five independent single-core serial jobs per ligand,
and each run involves a maximum of 10 cycles of MC/SA sampling of the ligand
within a 1 Å sphere. The center of the sphere is defined as the
center-of-geometry
of the input ligand pose. Each of cycles consists of 100 steps of MC
at high temperature followed by 1000 steps of SA towards a lower temperature.
At the beginning of each cycle, the ligand orientation/conformation will be
reset to the one found in the input file.
MC sampling moves are: i) molecular translation with a
maximum step size of 1 Å, ii) molecular rotation with a maximum step size of
180 degrees, and iii) intramolecular dihedral rotation with a maximum
step size of 180 degrees. For intramolecular dihedral rotation, only the
rotatable dihedral angles are selected for MC moves. The lowest LGFE scoring
pose from the MC sampling is used as starting pose in the following SA
sampling. SA sampling moves are smaller than for the MC phase: i) molecular
translation with a maximum step size of 0.2 Å, ii) molecular rotation with a
maximum step size of 9 degrees, iii) intramolecular dihedral rotation
with a maximum step size of 9 degrees. The lowest LGFE scoring pose from the
SA is saved in the multi-frame PDB file
3_silcsmc/<run>/pdb/<lig>_mc.<run>.pdb.
Best-pose retrieval¶
Once the SILCS-MC simulation is finished, retrieve the LGFE scores for each ligand subjected to SILCS-MC using:
${SILCSBIODIR}/silcs-mc/2_calc_lgfe_min_avg_sd ligdir=<directory containing lig mol2>
Note
If you used sdfile=<path to sdfile> option in the previous step, use
sdfile=<path to sdfile> in this step as well, instead of ligdir
option.
An example of the output of this script is:
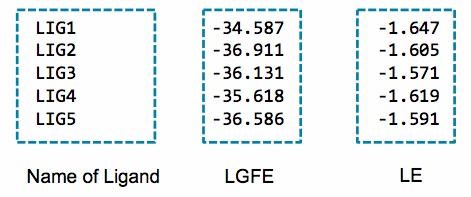
An alternative to the LGFE score is the ligand efficiency (LE). The LE is calculated as the LGFE score divided by the number of heavy atoms in each ligand.
User-defined protocols¶
In addition to the default docking and pose refinement protocols, users
can define their own SILCS-MC protocols. To do so, copy
${SILCSBIODIR}/templates/silcs-mc/params_custom.tmpl to the location where
you intend to run your custom SILCS-MC protocol. Edit this copy to reflect your
customization; see below for a detailed description of user-definable
parameters. Parameter values in angle brackets in this file, such as
<SILCSBIODIR>, will be replaced automatically at runtime where possible.
After you have edited params_custom.tmpl, use the following command to set
up and run SILCS-MC. Each input ligand can be stored as a separate SDF or Mol2
file. Alternatively, all input ligands can be combined in a single SDF file.
${SILCSBIODIR}/silcs-mc/1_run_silcsmc_custom prot=<prot pdb> \
ligdir=<directory containing ligand sdf/mol2> \
mapsdir=<directory containing SILCS FragMaps> \
paramsfile=<params_custom.tmpl>
The number of runs that will be spawned can also modified with the command-line
parameter totruns:
${SILCSBIODIR}/silcs-mc/1_run_silcsmc_custom prot=<prot pdb> \
ligdir=<directory containing ligand sdf/mol2>
mapsdir=<directory containing SILCS FragMaps>
paramsfile=<params_custom.tmpl>
totruns=<# of runs>
The full list of user-definable parameters for params_custom.tmpl is:
CGENFF_RULES <cgenff rules_file>(required)This file is needed by the internal CGenFF library to determine the correct force-field parameters for the ligand. The default value is
${SILCSBIODIR}/data/cgenff.rulesCGENFF_PAR <cgenff parameter file>(required)Along with the CGENFF_RULES file, this file is needed by the internal CGenFF library to determine the correct force-field parameters for the ligand. The default value is
${SILCSBIODIR}/data/par_all36_cgenff.prmSILCS_RULES <silcs rules file>(required)This rule file is used to map the different atoms in the ligand to the corresponding SILCS FragMap types. This mapping is used to determine the appropriate “field” that will be applied to the different atoms in the ligand when attempting an MC-move. The default value is
${SILCSBIODIR}/data/silcs_classification_rules_2018_generic_apolar_scale_1c.datAnother variant of this rule file is available at
${SILCSBIODIR}/data/silcs_classification_rules_2018_specific_standard_1c.dat.When
silcs_classification_rules_2018_generic_apolar_scale_1c.datis used, ligand atoms are assigned using six generic classifications in mapping them back to the FragMaps:APOLAR,HBDON,HBACC,MEOO,ACEO, andMAMN. All the atoms in the ligand fall into one of these six categories.When
silcs_classification_rules_2018_specific_standard_1c.datis used, ligand atoms are assigned using 13 more specific classifications in mapping them back to FragMaps:BENC,PRPC,AALO,MEOO,FORN,FORO,IMIN,IMIH,ACEO,MAMN,APOLAR,HBDONandHBACC. For instance, aromatic ring carbons and aliphatic linker carbons are distinguished asBENCandPRPC, respectively here, while they are grouped asAPOLARatoms in the generic rule file.GFE_CAP <default: 3.0>Maximum unfavorable GFE (kcal/mol) accounted in the MC calculation.
RDIE <default: true>When true, the distance dependent dielectric (RDIE) scheme is used to treat intramolecular electrostatics. When false, CDIE (constant dielectric scheme) is used.
DIELEC_CONST <default: 4>Dielectric constant used in the intramolecular electrostatic interactions calculations.
MIN_STEPS <default: 10000>Maximum number of steps of minimization performed using the steepest-descent algorithm with the ligand, before initiating MC simulation.
EMTOL <default: 0.01>Minimization is converged when the diff in total energy (totE) across the last 10 steps is smaller than this value. Once this criteria is satisfied minimization terminates.
MC_MOVE_RANGE <default:1.0 180.0 180.0>.Maximum range of translation, rigid body rotation and dihedral rotation per step of MC simulation.
MC_PRNT_FRQ <default: 0>Number of intermediate steps of MC to be written into OUTMCPDBFILE.
MC_STEPS <default: 10000>Number of steps of MC simulation to be performed per cycle.
SIM_ANNEAL_MOVE_RANGE <default:0.2 9.0 9.0>Maximum range of translation, rigid body rotation and dihedral rotation per step of simulated annealing simulation after MC simulation.
SIM_ANNEAL_STEPS <default: 40000>Number of steps of simulated annealing to be performed per cycle.
INIT_RUNS <default: 50>Number of MC/SA cycles before initiating checks for convergence.
NUM_TOL <default: 3>Number of top-scoring cycles with differences in LGFE less than DELTAE_TOLERANCE, before this simulation (run) is considered converged.
DELTAE_TOLERANCE <default: 0.5>When differences in LGFE of NUM_TOL most-favorable cycles are less than this defined tolerance value, convergence is reached and the program exits
DELTAE_BUFF <default: 10>Progression of MC+SA from one cycle to next is such that LGFE (of lowest conf) from MC should be less than (prev_min+deltae_buff). This ensures that N cycles are proceeding towards a minimum lower than that previously discovered lowest energy conformation.
TOTE_CRITERIA <default: false>When true, instead of LGFE, total energy (totE) of the system is used for convergence checks. Useful when running vacuum-phase MC simulations of the ligand.
TOT_RUNS <default: 250>Maximum number of MC simulation cycles. The program terminates if the
DELTAE_TOLERANCEcriteria is satisfied before reachingTOT_RUNS. Alternately, even if theDELTAE_TOLERANCEcriteria is not satisfied when the number of cycles executed reachesTOT_RUNS, the program terminates.RANDOM_SEED: <default: system-time>Seed used in MC simulation. When not set, system-time is used as a seed.
SIMULATION_CENTER: <x,y,z>Cartesian coordinates of where the MC simulation should be performed.
SIMULATION_RADIUS: <default: 10.0 A>Radius of the sphere within which MC simulation will be performed.
RANDOM_INIT_ORIENT: <true/false>When set to
TRUE,SIMULATION_CENTERshould also be set. The ligand is placed within a sphere of size,SIMULATION_RADIUS, in a random orientation and a conformationWhen set to
FALSE, then the center-of-geometry of the ligand is used as the center for the MC simulation. This is useful when the ligand pose in the pocket is well known.ATOM_TO_RESTRAIN: <atom number in sdf/mol2>When set, a spherical potential is applied to restrain the defined atom within the sphere during MC moves. This enables geometrically restraining a particular pharmacophore feature. Note, when using this feature, supply the full molecule with explicit hydrogens already added. Also, random pocket pose and placement using
RANDOM_INIT_ORIENTtrue is incompatible.When not set, the entire molecule is free to rotate/move/translate
ATOM_RESTRAINT_CENTER: <x,y,z>To be used in conjunction with
ATOM_TO_RESTRAINoption. This value is used to defined the center of the spherical potential.ATOM_RESTRAINT_RADIUS: <default: 1.0 A>To be used in conjunction with
ATOM_TO_RESTRAINoption. This value is used to defined the radius of the spherical potential. When not defined, then a default of 1 A is used.OUTRMSDFILE <output RMSD file>This file stores the RMSD and LGFE of the lowest energy conformation from each run of the MC/SA simulation. To be used in conjunction with
RANDOM_INIT_ORIENTset totrue.LIGAND_SDF: <ligand sdf file>When ligands are in SDF file format, use this option. If no input SDF file name is supplied at the command line, this line will be read.
LIGAND_MOL2: <ligand mol2 file>When ligands are in Mol2 file format, use this option. If no input Mol2 file name is supplied at the command line, this line will be read.
SILCSMAP <MapType> <map name>(required)Multiple
SILCSMAPflags can be defined, with each flag pointing to one file. Standard SILCS FragMaps of the following<MapType>should be included with the below keywords:EXCL - exclusion map
NCLA - zero map for non-classified ligand atoms
BENC - benzene carbon
PRPC - propane carbon
MEOO - methanol alcohol oxygen
FORN - formamide nitrogen
FORO - formamide oxygen
MAMN - methyl ammonium nitrogen
ACEO - acetate oxygens
AALO - acetaldehyde oxygen
IMINH - imidazole donor nitrogen
IMIN - imidazole acceptor nitrogen
APOLAR - generic non polar : green
HBDON - generic donor: blue
HBACC - generic acceptor: red
OUTMCPDBFILE <output PDB file>(required)This file stores the lowest energy conformation from each cycle of the MC/SA simulation.
OUTMCLOGFILE <output log file>This file stores the energy statistics of the lowest energy conformation from each cycle of the MC/SA simulation.
OUTMCPDBFILEPREFIX <output PDB file(s)>When reading SDF files (with one or more molecules), this file(s) stores the lowest energy conformation from each cycle of the MC/SA simulation. Filenames are appended with the name of the ligand(s).
OUTMCLOGFILEPREFIX <output log file(s)>When reading SDF files (with one or more molecules), this file(s) stores the energy statistics of the lowest energy conformation from each run of the MC/SA simulation. Filenames are appended with the name of the ligand(s).
If
LIGAND_SDFis defined, thenOUTMCPDBFILEPREFIXandOUTMCLOGFILEPREFIXneed to be defined and need to point to the right files for SILCS-MC to proceed.Note
Ligand related parameters such as
LIGAND_MOL2/LIGAND_SDF,OUTMCPDBFILE/OUTMCLOGFILE, need not be manually set. The script1_run_silcsmc_customwill set these parameters appropriately for each ligand in theligdirdirectory.