SILCS-Hotspots: Fragment Binding Sites Including Allosteric Sites¶
Background¶
SILCS-Hotspots identifies all potential fragment binding sites on a target by doing SILCS-MC sampling across the entire target structure. The protein or other macromolecular target is partitioned into a collection of subspaces in which the fragment is randomly positioned and subjected to extensive SILCS-MC to identify favored local poses. This may be performed 1000 times or more per fragment in each subspace. The identified fragment positions are then RMSD-clustered followed by selection of the lowest energy pose in each cluster to define a binding site for further analysis.
SILCS-Hotspots may be applied to small fragment-like molecules as well as larger drug-like molecules. When multiple fragments are used, a second round of clustering may be performed over the different fragment types to identify binding sites occuppied by one or more of the fragment types included in the calculation.
Results from SILCS-Hotspots include all potential binding poses of the individual fragments, binding sites that contain one or more of the fragment types, and ranking of fragment poses and binding sites based on LGFE scores. Applications of SILCS-Hotspots include identifying putative fragment binding sites and performing fragment-based drug design. Putative binding sites identified by SILCS-Hotspots can be used for rapid database screening with SILCS-Pharmacophore. Alternatively, sites identified by SILCS-Hotspots can be considered for fragment-based design by linking poses of fragment-like molecules in adjacent sites to create drug-like molecules.
Usage and options¶
Running SILCS-Hotspots calculations¶
Inputs for SILCS-Hotspots are the protein or other macromolecular target used for the SILCS simulations, the SILCS FragMaps resulting from those simulations, and the fragment(s) to be used for sampling. SilcsBio provides fragment files appropriate for SILCS-Hotspots (see below). Alternatively, you may use your own fragment file(s).
$SILCSBIODIR/silcs-hotspots/1_setup_silcs_hotspots prot=<prot PDB> ligdir=<fragment database> mapsdir=<mapsdir> bundle=<true/false>
The input arguments are the PDB file used for the SILCS run (prot), and the
locations of the fragment files (ligdir) and SILCS FragMaps (mapsdir).
When SILCS-Hotspots calculations are performed, the subdirectory 4_hotspots/
is created to store output from the exhaustive SILCS-MC runs.
Note
The above command will submit a large number of jobs, which can strain
job queueing systems. The bundle keyword launches bundled jobs instead
of individual jobs.
Options include:
FragMap directory path:
mapsdir=<location and name of directory containing FragMaps; default=maps>
By default, the program looks for FragMaps in the
maps/directory.Fragment database location:
ligdir=<location and name of directory containing fragment mol2/sdf>
You may prepare your own directory with Mol2/SDF files. There are also three directories of fragment-like molecules provided with your SilcsBio software:
Number of processors to use for bundled jobs:
nproc=<# of processors used when bundle=true>
Directory containing output to be used in subsequent steps:
hotspotsdir=<location of hotspots data; default=4_hotspots>
Template file for SILCS-MC sampling:
paramsfile=<custom params file>
The default SILCS-MC job parameters file for SILCS-Hotspots is
$SILCSBIODIR/templates/silcs-hotspots/params.tmpl. See User-defined protocols for customization details.
Post-run clustering¶
After all the SILCS-Hotspots jobs are complete, the next step is to cluster fragment poses that were output by the completed jobs. The clustering algorithm iteratively finds clusters with the largest number of members [13]. The process entails 1) computing the number of neighbors of each pose, 2) choosing the pose with the largest number of neighbors and marking it and its neighbors as cluster members, 3) removing cluster members identified in Step 2 from the pool of poses, and 4) repeating Steps 1-3 until there are no available poses left. The cluster “center” is defined as that fragment pose with the most neighbors in that cluster. This pose’s LGFE score defines the LGFE score of the cluster. Run the command
$SILCSBIODIR/silcs-hotspots/2_collect_hotspots prot=<prot PDB> ligdir=<fragment database>
to perform the clustering. You may also specify the following additional options:
Radius for RMSD clustering:
cutoff=<cutoff for clustering; default=3>
Maximum number of sites to be identified for each fragment:
maxsites=<maximum number of sites ordered by LGFE; default=200>>
Directory containing output from the previous steps:
hotspotsdir=<location of hotspots data; default=4_hotspots>
Note
2_collect_hotspots and 3_create_report have external Python dependencies. You may need to install the following Python
packages (for example by using pip install <package name>):
numpy
scipy
tqdm
pyyaml
xlsxwriter
Site determination and report generation¶
The final step of SILCS-Hotspots is site determination and report generation.
This involves a second round of clustering over all specified fragments to
identify sites on the protein to which one or more fragments bind. Each site is
given the average LGFE score over all the fragments. In addition, the sites
themselves may be clustered to identify binding pockets that suggest multiple
adjacent sites that may be linked to build larger fragments (under development).
Information on all the fragments, the binding sites, and the binding pockets is
included in report.xlsx and in PDB files that are output to the
subdirectory 4_hotspots/report/. Run the following command
$SILCSBIODIR/silcs-hotspots/3_create_report ligdir=<fragment database>
You may add these options:
Directory containing output from the previous steps:
hotspotsdir=<location of hotspots data; default=4_hotspots>
Cluster radius for binding site determination:
site_cutoff=<cluster radius for site determination; default=6.0>
LGFE cutoff for inclusion of fragment poses in site determination:
ligand_lgfe_cutoff=<Ligand LGFE cutoff for site determination; default=-2.0>
Average LGFE cutoff for site determination:
site_lgfe_cutoff=<average LGFE cutoff for binding sites; default=-2.0>
Sites having values less favorable than the cutoff will be discarded.
Flag to activate binding pocket calculation and associated clustering radius (under development):
pocket=<perform pocket analysis; default=False> pocket_cutoff=<cluster radius for binding pocket determination; default=12.0>
The 3_create_report command will populate the 4_hotspots/report/
subdirectory with the following:
report_all.xlsx: Spreadsheet file containing analysis information. Included are the LGFE and Ligand Efficiency (LE) scores for each copy of every fragment obtained from clustering, relative affinity analysis, and listing of the posed fragment that defines each site. If binding pocket analysis is performed, information on binding pockets is included.hotspots_sites.pdb: PDB file containing the identified sites. The B-factor column value includes the average LGFE score of that site.pdb_by_ligands: Directory containing PDB files for each fragment with multiple coordinates for each site identified for the fragment. In the PDB files, REMARK includes the LGFE scores, the B-factor column includes the GFE score for each atom, and the final column lists the SILCS atom type.pdb_by_site: Directory containing PDB files of the fragments located at each site inhotspots_sites.pdb. For example, the filenamesite_all_1_8_1.pdbindicates that at site 1 fragment 8 is present. The final 1 indicates that this is the first copy of fragment 8 at site 1. From the clustering alogrithm, it is possible that more than one copy of a fragment is included in a site. For example,site_all_1_8_2.pdbwould be the second copy of fragment 8. In each PDB file, REMARK includes the LGFE scores, the B-factor column includes the GFE score for each atom, and the final column lists the SILCS atom type.
The following additional outputs are produced if binding pocket
analysis is performed (pocket=true):
hotspots_pockets.pdb: PDB file containing sites that define each binding pocket. In the following, pocket P01 or 1 is defined by four sites with LGFE values for each site shown in the B-factor column. The algorithm does not number pockets consecutively: in this example, there is no P02 pocket, and pocket P03 contains 2 sites.ATOM 1 X P01 A 1 20.980 7.230 -12.513 1.00 -4.39 C ATOM 2 X P01 A 1 27.947 16.848 -6.986 1.00 -2.82 C ATOM 3 X P01 A 1 17.606 12.224 -10.043 1.00 -2.67 C ATOM 4 X P01 A 1 14.912 17.557 -5.282 1.00 -2.08 C ATOM 5 X P03 A 3 16.182 -10.455 11.793 1.00 -2.96 C ATOM 6 X P03 A 3 17.605 0.058 15.384 1.00 -2.83 C
pdb_by_pocket: Directory containing PDB files for fragments that comprise each pocket. For example,pocket_all_8_site_12_10_1.pdbindicates pocket 8 contains a fragment from site 12 and that fragment is fragment 10.1indicates that it is the first copy of fragment 10 in that pocket. REMARK includes the LGFE scores, the B-factor column includes the GFE score for each atom, and the final column is the SILCS atom type.
Practical considerations¶
1_setup_silcs_hotspots
creates a subdirectory, 4_hotspots/, (or user defined name using hotspotsdir=) that contains fragment pose and
LGFE information. As SILCS-Hotspots makes use of
large numbers of SILCS-MC calculations on each fragment, this directory
will be filled with a substantial amount of data. It is suggested that, once
all analyses are complete, these files either be deleted or archived,
and 4_hotspots/ be renamed prior to additional
hotspots runs. Remember, the data in 4_hotspots/ are used for
post-run clustering and site determination and report generation.
If 1_setup_silcs_hotspots is being rerun with new
parameters, such as new fragments as specified by ligdir=, the subdirectory
4_hotspots/ should be renamed or an alternate name assigend using hotspotsdir=
to avoid information from the original
run being overwritten. However, this is not strictly necessary IF all
the new fragments have unique filenames relative to the original run AND
the SILCS-MC job parameters specified by paramsfile= are not changed,
since the subsequent 2_collect_hotspots command only performs analysis on
Mol2/SDF files in the specified ligdir directory.
1_setup_silcs_hotspots launches a large number of jobs that, while
the majority of jobs finish quickly individual jobs may take time (minutes to an hour) to complete.
In some cases one or two jobs in the set may require additional time due
to the robust SILCS-MC convergence criteria used in SILCS-Hotspots.
For the SILCS-MC sampling, especially with larger fragments or ligands, you may prefer not
to include the SILCS exclusion map. This will permit fragment sampling of
poses that would otherwise be rejected because of overlap with the
exclusion region. Using this approach, final fragment
poses will be based solely on scoring with SILCS FragMaps. To achieve
this, set the weighting of the exclusion map to zero in your custom
SILCS-MC job parameters file (paramsfile=<custom params file>).
A simple means to this end is to use a copy of
the default file $SILCSBIODIR/silcs-mc/params_custom.tmpl in which you
replace the 1.000 in the below line with 0.000.
SILCSMAP EXCL <MAPDIR>/<prot>.excl.map 1.000
Clustering of data in the 4_hotspots/ or equivalent subdirectory with the
2_collect_hotspots command produces representative fragment poses on a
per-cluster basis. If clustering is repeated, for example with a different
clustering radius, the old PDB files created in 4_hotspots/ will be
overwritten. Therefore, make sure to run site determination and report
generation (the 3_create_report command) on your original
clustering output before repeating clustering.
2_collect_hotspots processes each of the fragments individually and
typically requires minutes to complete for 100 fragments. During that process a
number of warning messages, such as “Clustered PDB not found:
4_hotspots/2/subspace_1/pdb/2_clust_1_1.pdb,” will be given. These are expected
as they indicate subspaces for the fragments in which no favorable fragments poses
were identified. For example, this may occur where the subspace encompasses
the protein structure.
Site determination and report generation with the 3_create_report command
creates a subdirectory,
4_hotspots/report/, and outputs report.xls and PDB files into this
subdirectory. Rerunning 3_create_report with different parameters,
like an alternate value of the site clustering cutoff, will overwrite the
original output. Therefore, prior to rerunning report generation, rename
4_hotspots/report/ to save your original report generation outputs.
The clustering alogrithm for site determination does not consider the
identity of the fragment when performing the clustering. Accordingly, in certain
cases it is possible for the same fragment to be included two or more times in a
given site.
Example¶
The following demonstrates use of SILCS-Hotspots on p38 MAP kinase.
Input files, including FragMaps, are in $SILCSBIODIR/examples/silcs/.
cp -r $SILCSBIODIR/examples/silcs/silcs_fragmaps_p38a .
cd silcs_fragmaps_p38a
$SILCSBIODIR/silcs-hotspots/1_setup_silcs_hotspots prot=p38a.pdb ligdir=$SILCSBIODIR/data/databases/ring_subset mapsdir=maps bundle=true
In this example, we use $SILCSBIODIR/data/databases/ring_subset as the
fragment database. Owing to the small size of this database, the example run
finishes quickly. Below are the ring fragments in the ring_subset
database.
Once the jobs spawned by 1_setup_silcs_hotspots complete, use the
following commands for post-run clustering and site determination and
report generation.
$SILCSBIODIR/silcs-hotspots/2_collect_hotspots prot=p38a.pdb ligdir=$SILCSBIODIR/data/databases/ring_subset
$SILCSBIODIR/silcs-hotspots/3_create_report ligdir=$SILCSBIODIR/data/databases/ring_subset
This will have created the 4_hotspots/report/ subdirectory containing the
following:
hotspots_sites.pdb: Centroid positions of fragment clusters, that is, the “hotspots.” Clusters are ranked using average LGFE scores, with the cluster rank listed in the residue number field and the average LGFE score in the B-factor field.report_all.xlsx: Report of hotspots in spreadsheet format.pdbs_by_sites: List of PDB files for each hotspot.
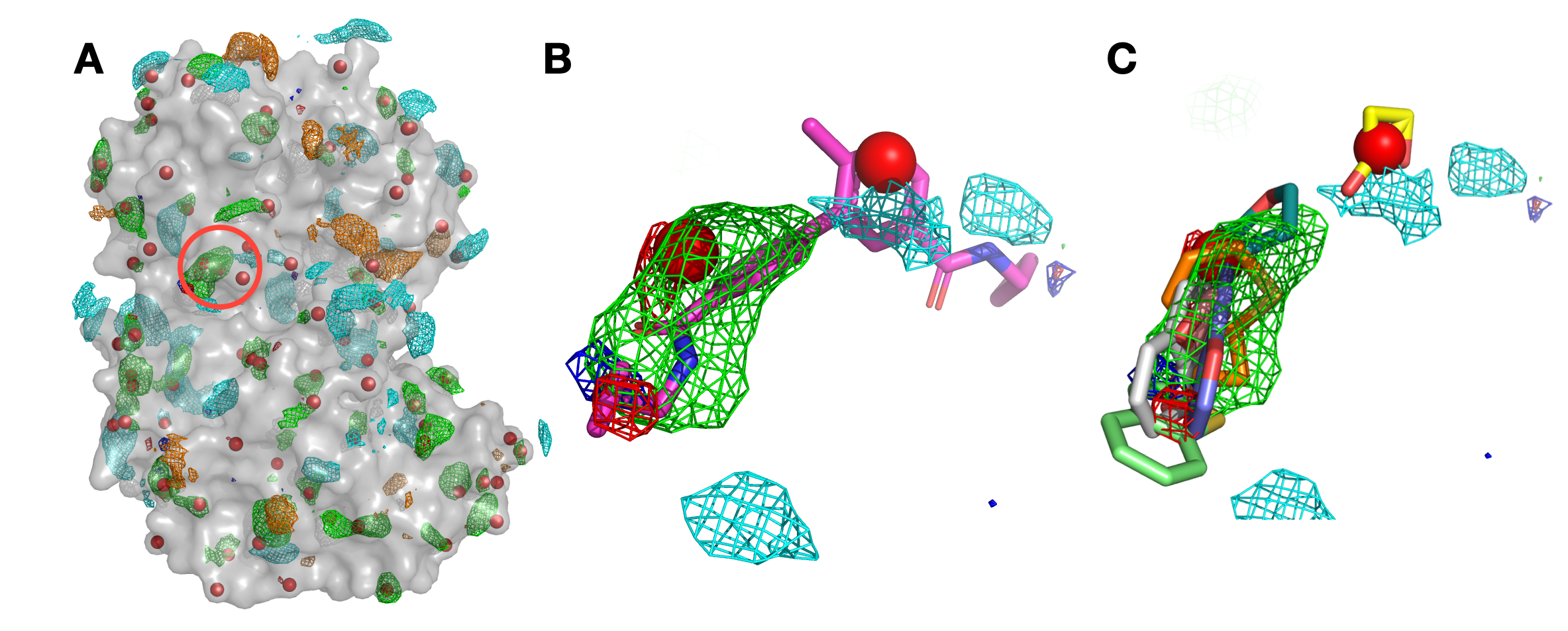
Panel (A) shows p38 (surface representation), SILCS FragMaps (wire frame), and all hotspots (red spheres) determined by SILCS-Hotspots. The crystal binding pocket is circled in red. Panel (B) has FragMaps, hotspots, and the crystallographic ligand. Two hotspots are identified within the crystal binding pocket and coincide with the rings of the fragment. Panel (C) shows fragment poses from SILCS-Hotspots calculations as well as FragMaps and hotspots.