CGenFF: CHARMM General Force Field¶
Background¶
The CHARMM General Force Field (CGenFF) covers a wide range of chemical groups present in biomolecules and drug-like molecules, including a large number of heterocyclic scaffolds [3]. The parametrization philosophy behind the force field focuses on quality at the expense of transferability, with the implementation concentrating on an extensible force field.
CGenFF uses the CHARMM additive potential energy function to calculate the energy as a function of the Cartesian coordinates of the system, as shown below.
The bonded or intramolecular part of the potential energy function consists of terms for the bonds, valence angles, torsion or dihedral angles, improper dihedral angles, and a Urey-Bradley term, where \(b_0\), \(\theta_0\), \(\phi_0\), and \(r_{1,3;0}\), respectively, are the bond, angle, improper and Urey-Bradley equilibrium values, the \(K\)’s are the corresponding force constants, and \(n\) and \(\delta\) are the dihedral multiplicity and phase. The nonbonded or intermolecular portion consists of an electrostatic term, with \(q_i\) and \(q_j\) being the respective partial atomic charges on atoms \(i\) and \(j\), and a van der Waals (vdW) term, which is treated by the Lennard-Jones (LJ) 6-12 potential in which \(\epsilon_{ij}\) is the well depth, \(R_{\mathrm{min},ij}\) is the radius, and \(r_{ij}\) is the distance between \(i\) and \(j\).
It is apparent that a simulation on any system of practical interest requires large numbers of parameters. To make the assignment of these parameters practical, force fields require atom types to be assigned to all the atoms in the system, with the parameters associated with combinations of atom types. For instance, the parameter list will contain \(K_\phi\), \(n\), and \(\delta\) values for the dihedral parameters associated with all combinations of four atom types that occur in the molecules supported by the force field. Thus, the first step of assigning parameters for a chemical system is assigning atom types to that system.
CGenFF comes with an algorithm that can automatically assign atom types to a given molecule. The atom typer is rule-based and programmable, making it easy to implement complex atom typing rules and to update the atom typing scheme as the force field grows. Assignment of bonded parameters is based on substituting atom types in the definition of the desired parameter. A penalty is associated with every substitution and the existing parameter with the lowest total penalty is chosen as an approximation for the desired parameter. Charges are assigned using an extended bond-charge increment scheme that is able to capture inductive and mesomeric effects across up to 3 bonds. More details can be found in [4].
Usage¶
The CGenFF program accepts Mol2 format files. For correct atom typing, it is important that all hydrogens are present, the system has correct protonation and tautomeric states, and that the bond orders are correct.
Once the Mol2 file is ready, the CGenFF program can be executed:
${SILCSBIODIR}/programs/cgenff <mol2file>
This produces a stream file in standard output. The output can be redirected to
a .str file. The stream file consists of a topology file and the extra
parameter file combined in a single outpu. It is up to the user to whether to
use the stream file directly or to split the file into two separate files.
As an example, the adrenalin molecule is parametrized in CGenFF. The example
input and output files can be found in
${SILCSBIO}/examples/cgenff/adrenalin.mol2 and
${SILCSBIO}/examples/cgenff/adrenalin.str, respectively.
The output stream file has two major sections.
* Toppar stream file generated by
* CHARMM General Force Field (CGenFF) program version 1.0.0
* For use with CGenFF version 3.0.1
*
read rtf card append
... topology section ...
END
read param card flex append
... parameter section ...
END
RETURN
In CHARMM, asterisk and exclamation marks represents comments. If you wish to
separate topology and parameter sections, you can take out the corresponding
section after read rtf or read param to the END statement.
Let’s take a look at topology section:
RESI ADRN 0.000 ! param penalty= 113.000 ; charge penalty= 16.607
GROUP ! CHARGE CH_PENALTY
ATOM O1 OG311 -0.649 ! 11.757
ATOM O2 OG311 -0.530 ! 0.000
ATOM O3 OG311 -0.530 ! 0.000
ATOM N NG311 -0.524 ! 16.607
ATOM C1 CG311 0.127 ! 16.171
Above is the beginning of the topology section. This section defines the atom names and types, as well as the atomic charges. Each entry is followed by a charge penalty score. The atomic partial charges are assigned based on chemical analogy to existing parametrizations in the force field. The penalty score becomes high when a good analogy is not found.
Below is an excerpt from the parameter section. When an exact parameter is not found in an existing parameter database, an entry is added based on an analogous parameter. Similar to atomic partial charges, each entry in the parameter section is also followed by a penalty score.
ANGLES
CG2R61 CG311 OG311 75.70 110.10 ! ADRN , from CG2R61 CG321 OG311, penalty= 4
CG311 CG321 NG311 43.70 112.20 ! ADRN , from CG331 CG321 NG311, penalty= 1.5
CG321 NG311 CG331 40.50 112.20 5.00 2.42170 ! ADRN , from CG3AM1 NG311 CG3AM1, penalty= 35
High penalty scores indicate that a parameter may be targeted for further optimization. Typically, penalty scores greater than 50 warrant further optimization.
It is possible to request that the CGenFF program preserve atomic partial
charges from the input Mol2 file instead of assigning these values. This is
done using the -z flag:
${SILCSBIODIR}/programs/cgenff -z <mol2file>
Warning
We STRONGLY advise against using your own custom atomic partial charges.
Nonbonded interactions in the force field depend upon both the Coulomb and
the Lennard-Jones terms, and the parameter values for these terms have been
carefully developed together. Assigning custom atomic partial charges
with the -z flag is likely to disrupt the balance in these nonbonded
force field terms.
GROMACS-readable parameters¶
To get parameters in GROMACS format, execute the following command.
${SILCSBIODIR}/cgenff/cgenff_to_gmx mol=<mol2file>
This command produces three files <mol2file>_gmx.top, <mol2file>_gmx.pdb & <mol2file>_charmm.str.
<mol2file>_gmx.top/pdb contain the GROMACS readable parameters and the coordinate information.
<mol2file>_charmm.str contain CHARMM readable parameters.
CGenFF Parameter Optimizer¶
The CGenFF program performs atom typing and assignment of parameters and charges by analogy in a fully-automated fashion. Atom typing is done by a deterministic programmable decision tree. Assignment of bonded parameters is based on substituting atom types in the definition of the desired parameter. A penalty is associated with every substitution and the existing parameter in the current version of the force field with lowest total penalty is chosen as an approximation for the desired parameter. The penalty score is returned to the user as an indication of the approximation needed to obtain the desired parameter. For example, dihedral parameters with higher penalties (typically >50) may be candidates for further optimization. When using the Optimizer, the atom typing and parameter assignment is performed internally such that the user only needs to supply the Mol2 file of the molecule of interest.
The threshold for selecting parameters for optimization is based on multiple criteria, including user preference. The penalty score is determined by the analogy of the atom types associated with the new parameter to those of parameters already in the CGenFF force field. Parameters with high penalties may actually be of satisfactory accuracy while parameters with low penalties may be of poor accuracy. Accordingly, the user should make an initial evaluation of the treatment of a given molecule by CGenFF and then identify the parameters that may contribute to possible inaccuracies in the conformational properties of the molecule or its interactions with the environment. Dihedral parameters will typically have the largest impact on the conformational properties of a molecule and, therefore, should be considered for optimization.
CGenFF Parameter Optimizer allows for automatic optimization of rotatable dihedrals. Once the user specifies the dihedral to be optimized, the Optimizer coordinates the generation of quantum mechanical (QM) target data followed by the fitting of force field parameters to these target data. The Optimizer will first spawn Psi4 QM jobs. It will then collate the resulting QM dihedral scan data. Finally, it will fit force field parameters to these data using the least-square fitting procedure LSFitPar and output the new, optimized force field parameters. If multiple dihedrals are to be optimized, then each dihedral is fitted separately targeting independent QM scans on each dihedral. During the fitting, the initial multiplicities are those assigned by the CGenFF program. Once the initial fit is complete and the RMS error determined, the program automatically refits the data using a multiplicity of 1, then multiplicities of 1 and 2, 1, 2 and 3 and 1, 2, 3 and 6. If improvements in the RMSE are obtained beyond a cutoff (default is 10% of the RMSE) with the additional multiplicities, then those parameters are selected.
The Optimizer accepts molecules in Mol2 file format, with the following requirements: hydrogens must be explicitly defined, ionizable groups must have correct protonation states, and bond-order between atoms must be correctly defined.
To run the program, set the following environment variables,
export SILCSBIODIR=<silcsbio>
export PSI4DIR=<psi4>
export GMXDIR=<gromacs/bin>
The Psi4 package can be obtained from the Psi4 download page. For the easiest installation, use the Psi4 Binary Installer, run the command:
./Psi4conda-latest-py36-Linux-x86_64.sh
and following the step-by-step guide.
After Psi4 has been installed, set the PSI4DIR variable correctly and
proceed with using the optimizer. To make sure PSI4DIR is correctly
set, check if $PSI4DIR/bin/psi4 is accessible.
Note that downloading the Psi4 source code and compiling the program locally with the appropriate switches can lead to a significant gain in performance over the downloaded binary version.
Usage¶
${SILCSBIODIR}/cgenff-optimizer/optimize_cgenff_par mol=<mol2file>
The second line of the Mol2 file contains a string naming the molecule. Certain molecular modeling programs will put the string “*” on this line instead of a descriptive name for the molecule. In such cases, please replace the string with an alphanumeric string, else the Optimizer will fail.
Along with the required argument of mol=<mol2file>, the following additional
parameters can also be set:
Penalty score :
penalty=<score; default 50>By default, the program identifies rotatable dihedrals with penalties greater than 50 for optimization. This can be changed by adjusting the
penaltyargument, to increase/decrease the list of dihedrals that will be optimized.
Dihedral step size :
dihedral_step_size=<step size; default 15>Each dihedral identified for optimization is rotated through 360 degrees with a step-size of 15 degrees to generate QM target data. This step-size can be changed by adjusting the
dihedral_step_sizeargument to increase or decrease the resolution of the dihedral scan.
Number of cores used for QM optimization:
nproc=<number of core; default all available cores on workstation/8 cores on HPC>At each scan point for each dihedral, a constrained QM optimization is performed. Psi4 can make use of multiple CPU cores to run this optimization more quickly. When the Optimizer is installed on a high-performance computing cluster (HPC), separate jobs are submitted for each of the identified dihedrals simultaneously, and each job requests
nproccores. When the Optimizer is installed on a workstation, then each scan point for each dihedral is run one after the other using all available cores on the workstation. This default behavior can be changed using thenprocargument.
Molecular-orbital theory:
mo_theory=<MP2/HF; default MP2>Constrained QM optimization for each dihedral at each scan point is performed using Møller–Plesset perturbation theory (MP2, specifically the Psi4 DF-MP2/6-31G(d) model chemistry). If the user wants to instead use Hartree-Fock (HF), then it can be set using
mo_theory.
Energy difference cutoff to eliminate rotamers from QM optimization and subsequent fitting:
dg_cutoff=<energy_difference; default 100.>The optimizer initially generates all the rotamers for each dihedral being optimized as required for the potential energy surface (PES) and performs a CGenFF energy optimization on each restrained rotamer. If the relative energy of each rotameter is greater than the
dg_cutoffvalue (kcal/mo), that rotatamer is omitted from the QM optimization and subsequent parameter fitting. This is performed to eliminate rotamers with steric clashes that can be problematic for the QM optimization as well as lead to the parameter optimizer attempting to fit to high energy regions of the PES that are typically not sampled in MD simulations.
Specification of compute node for job submission:
hpc=<true/false; default true>
Installation of the CGenFF optimizer specifies the computer on which the jobs will be run, which is typically a local HPC cluster such that the default is
hpc=true. However, if the user wants to run the job on their local workstation thenhpc=falsemay be specified. Alternatively, if the default is false,hpc=truecan be specified to run the job on the HPC cluster. This requires that the appropriate cluster be specified in the default installation.
Running jobs sequentially or in parallel when
hpc=true.jobtype=<seq/par; default seq>
When the QM jobs are submitted to an HPC queue they will run sequentially using the specified number of processors (
nproc). However, if adequate compute resources are available it is possible to have all the QM jobs run simultaneously in parallel by specifyingjobtype=parwith each job using the specified number of processors (nproc). Care should be taken when using this option as the number of individual QM jobs can be quite large (n x 24 jobs where n is the number of dihedral parameters being optimized). However, use of the option can lead to the QM jobs finishing rapidly.
Setting the RMS energy difference to not redo the least-squares fitting using different multiplicities.
rmse_cutoff=<RMS energy cutoff to determine if additional multiplicities should be tested; default=0.0>
The initial least-squares fit applies the multiplicities in the original CGenFF parameters assigned to the dihedral being optimized. From the fit the RMS difference between the target QM and optimized MM parameters is calculated. If that values is greater than
rmse_cutoffthen least-squares fitting is performed with additional multiplicities. rmse_cutoff is set to zero by default to force the additional multiplicities to be included in the least-squares fitting. Note that the fitting may be repeated using the calculated QM data as described in the ``Practical Considerations” section below.
Example Usecase¶
The following demonstrates usage of the CGenFF Parameter Optimizer. The input
file ethene_co_isoxazole.mol2 is available in
${SILCSBIODIR}/examples/cgenff/. When running the Optimizer it is suggested
that a subdirectory be created for each molecule and the job run from within
that subdirectory.
${SILCSBIODIR}/cgenff-optimizer/optimize_cgenff_par mol=ethene_co_isoxaole.mol2
The resulting output is a CHARMM-compatible CGenFF stream file named
ethene_co_isoxazole_optimized.str. Since this molecule does not contain any
dihedral parameter with penalty greater than 50, running the above command
results in:
Nothing to optimize based on the penalty score threshold of 50
The user can look through the output stream file to identify dihedral parameters that may benefit from optimization:
DIHEDRALS
CG2DC3 CG2DC1 CG2O5 CG2R51 1.4000 2 180.00 ! NONAME.* , from CG2DC3 CG2DC1 CG2O5 OG2D3, penalty= 26.5
HGA4 CG2DC1 CG2O5 CG2R51 0.0000 2 180.00 ! NONAME.* , from HGA4 CG2DC1 CG2O5 OG2D3, penalty= 26.5
CG2DC1 CG2O5 CG2R51 CG2R51 1.5850 2 180.00 ! NONAME.* , from CG2O3 CG2O5 CG2R61 CG2R61, penalty= 46.5
CG2DC1 CG2O5 CG2R51 OG2R50 1.5850 2 180.00 ! NONAME.* , from CG2O3 CG2O5 CG2R61 CG2R61, penalty= 49
OG2D3 CG2O5 CG2R51 CG2R51 1.5850 2 180.00 ! NONAME.* , from OG2D3 CG2O5 CG2R61 CG2R61, penalty= 33.5
OG2D3 CG2O5 CG2R51 OG2R50 1.5850 2 180.00 ! NONAME.* , from OG2D3 CG2O5 CG2R61 CG2R61, penalty= 13
CG2O5 CG2R51 CG2R51 CG2R52 0.0000 2 180.00 ! NONAME.* , from CG2O1 CG2R51 CG2R51 CG2R52, penalty= 3
CG2O5 CG2R51 CG2R51 HGR51 1.0000 2 180.00 ! NONAME.* , from CG2R51 CG2R51 CG2R51 HGR51, penalty= 16.5
CG2O5 CG2R51 OG2R50 NG2R50 8.5000 2 180.00 ! NONAME.* , from CG2R51 CG2R51 OG2R50 NG2R50, penalty= 16.5
If the user now wants to fit CG2DC1 CG2O5 CG2R51 OG2R50, which has a
penalty score of 49, the program can be re-run with a lowered penalty threshold
of 48:
${SILCSBIODIR}/cgenff-optimizer/optimize_cgenff_par mol=ethene_co_isoxazole.mol2 penalty=48
This will result in:
#############################################
for dihedral = CG2DC1 CG2O5 CG2R51 OG2R50, parameter penalty = 176.000000;
#############################################
Will be optimizing the parameters of the above listed dihedral(s)
Note that all parameters associated with dihedrals about a rotatable bond with a penalty score exceeding the penalty threshold will be optimized. Dihedrals that are considered rigid based on being located in rings, being double bonds and so on, are excluded from fitting. This selection process can lead to a situation in which two or more sets of dihedral parameters associated with the same rotatable bond are being fit independently. While each of those fits may appear to be successful, when those parameters are combined and applied to the molecules of interest, the conformational energy for rotation about that bond will likely be inaccurate. Such a situation should be avoided by setting the penalty tolerance to be higher that the penalty values for those parameters and one of the dihedrals selected for optimization as described in the following section.
If the user wants to fit the parameters for one or more specific dihedrals if none are selected based on the penalty score, or in addition to the automatically selected dihedrals, the user can manually supply the four atoms defined each specific dihedral on prompt:
${SILCSBIODIR}/cgenff=optimizer/optimize_cgenff_par mol=ethene_co_isoxazole.mol2 penalty=50
will result in :
CHARMM General Force Field (CGenFF) program version 2.1.0
released the 27th of October 2016
Copyright (C) 2017 SilcBio LLC
and University of Maryland, School of Pharmacy. All Rights Reserved.
Now processing molecule sulf ...
#############################################
#############################################
Nothing to optimize based on the penalty score threshold of 50
Do you want to perform optimization with anymore dihedrals? [Y/N; default N]
To add to the list, type Y, and then supply dihedral(s) with the format
AtomType1 AtomType2 AtomType3 AtomType4. The program then summarizes the
updated-list:
List updated; will be attempting optimization with the following dihedrals
#############################################
CG2DC1 CG2O5 CG2R51 OG2R50
#############################################
The last column of the PDB file <mol>_cgenff_atomtypes.pdb created by
the Optimizer identifies the atom type for each atom in the system which may be
inspected to select specific parameters for optimization.
For the current example, from the information in
ethene_co_isoxazole_cgenff_atomtypes.pdb, atom 5 has the atom type
CG2R51:
ATOM 1 C NONA 1 0.759 0.680 1.188 1.00 0.00 CG2R52
ATOM 2 H NONA 1 1.645 1.222 1.497 1.00 0.00 HGR52
ATOM 3 C NONA 1 -0.353 1.220 0.530 1.00 0.00 CG2R51
ATOM 4 H NONA 1 -0.508 2.244 0.227 1.00 0.00 HGR51
ATOM 5 C NONA 1 -1.205 0.177 0.349 1.00 0.00 CG2R51
ATOM 6 C NONA 1 -2.542 0.163 -0.296 1.00 0.00 CG2O5
ATOM 7 C NONA 1 -3.305 -1.129 -0.378 1.00 0.00 CG2DC1
ATOM 8 H NONA 1 -2.855 -2.041 0.043 1.00 0.00 HGA4
ATOM 9 C2 NONA 1 -4.530 -1.255 -0.944 1.00 0.00 CG2DC3
ATOM 10 H21 NONA 1 -5.050 -2.226 -0.980 1.00 0.00 HGA5
ATOM 11 H22 NONA 1 -5.045 -0.383 -1.387 1.00 0.00 HGA5
ATOM 12 O1 NONA 1 -3.016 1.204 -0.756 1.00 0.00 OG2D3
ATOM 13 O NONA 1 -0.636 -0.949 0.875 1.00 0.00 OG2R50
ATOM 14 N NONA 1 0.613 -0.617 1.406 1.00 0.00 NG2R50
Following the identification of the dihedrals to the fitted, the Optimizer performs a complete molecular mechanics (MM) minimization of the molecule using the default CGenFF force field parameters. Next, each identified dihedral is independently rotated through 360 degrees with the specified step-size. Separate Psi4 jobs for constrained QM optimization around the specified dihedral are then performed either serially, one-by-one when running on a workstation, or in parallel when running on a HPC cluster.
The program waits on the QM jobs to finish. After the completion of the QM jobs,
the MM energy is computed for each scan point. Both the QM and MM energies are
then used by the lsfitpar program to fit the dihedral parameter. Updated
parameters are output as <mol>_optimized.str.
Graphs of the dihedral scan energies are output in the files files
qm_mm_*.png. If the computing system where you are running the Optimizer
does not contain gnuplot or gnuplot linked against the png library, you may copy
the 4plot directory to a computing system that does and run the gnuplot
files there.
Note that the most robust test of the optimized parameters involves testing them directly in a molecular modeling package with the new parameters added to the CGenFF parameter file (see below).
The following is a graph produced by the Optimizer for the
ethene_co_isoxazole example and demonstrates the better agreement with the
QM data following dihedral parameter optimization:
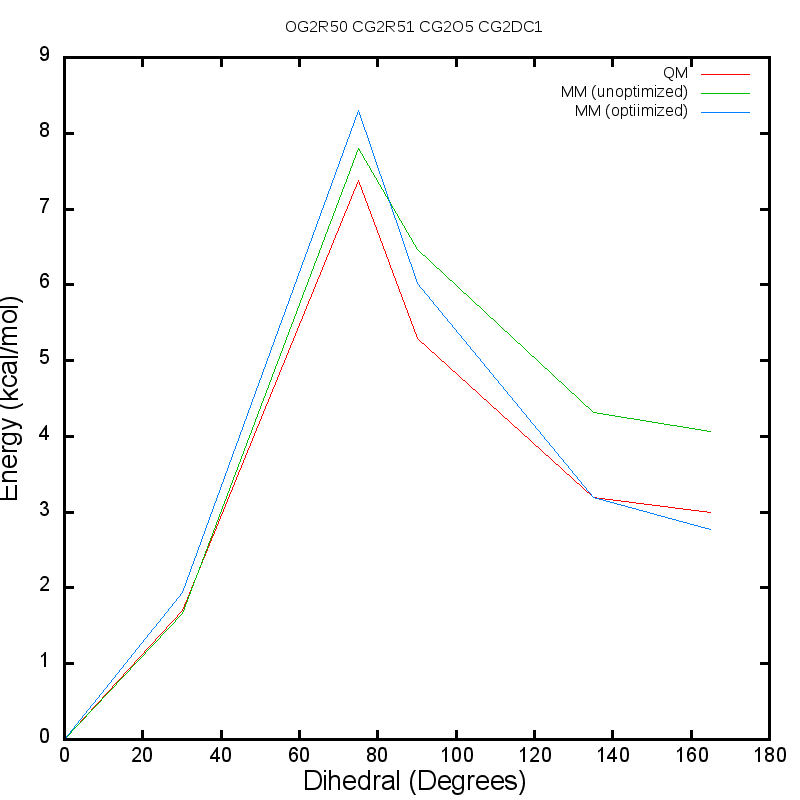
The default CGenFF parameter for dihedral CG2DC1 CG2O5 CG2R51 OG2R50 was
CG2DC1 CG2O5 CG2R51 OG2R50 1.5850 2 180.00 ! RMSE = 0.468999
and the replacement parameter produced by the Optimizer is
CG2DC1 CG2O5 CG2R51 OG2R50 0.5728 2 0.00 ! RMSE = 0.111323
Performing dihedrals optimization in the background¶
The above example runs the optimization on the command line requiring the terminal in which the job is submitted to remain open. To run the optimization in the background, the ‘nohup’ command may be used as in the following example. Note that a small script would be required to identify specific dihedrals for optimization.
nohup $SILCSBIODIR/cgenff-optimizer/optimize_cgenff_par.sh mol=ethene_co_isoxazole.mol2 > cgenff_opt_run1.txt &
Including new dihedral parameters with CGenFF¶
Currently, the optimized parameters generated by the CGenFF optimizer are NOT automatically added to the CGenFF parameter database. Accordingly, the user needs to manually add the parameters to the file using a standard editor (e.g., vi or emacs).
$SILCSBIODIR/data/par_all36_cgenff.prm
In the editor go the the DIHEDRALS section of the parameter file and then manually add the new parameters into the file. The new parameters are in filename_optimized.str found in the directory in which the optimization was run. “filename” corresponds to the molecule filehame.mol2 used to initiate the optimization. The optimized dihedral are at the end of the DIHEDRALS section of that file and are indicated by the comment containing the RMSE values obtained from the fitting of each dihedral parameter as follows.
CG2R51 CG2R51 NG311 CG331 3.8499 2 180.00 ! RMSE = 0.58618
CG2R51 CG2R51 NG311 CG331 1.5145 3 180.00 ! RMSE = 0.58618
CG3C52 CG2R51 NG311 CG331 2.1370 2 180.00 ! RMSE = 0.245345
CG3C52 CG2R51 NG311 CG331 1.4058 3 180.00 ! RMSE = 0.245345
Those parameters may be cut and paste directly into par_all36_cgenff.prm. Note that the file is generally protected and system administrator assistance is required to update this file. In the case of replacing dihedral parameters that are already in par_all36_cgenff.prm, the original parameters should be commented with a ! and the new parameters added.
Additional output information¶
Upon completion of the optimization the data generated is organized and placed in the subdirectory “raw_data” in which there is an additional subdirectory “psi4_calc” that contains files from the QM calculations.
optimize_cgenff_par.logSummary of the dihedrals being optimized and the optimization process including the RMSE data and final parameters.
dih_params_to_optimize*.txtDihedral parameters initially selected and updated list targeted for optimization.
params*.{inp,out}Inputs and outputs of MM minimizations to calculate the potential energy surfaces. Includes information on highly unfavorable conformations that were not included in the final parameter fitting.
params_aft_qm.outOutputs of MM minimizations using the optimized parameters to calculate the potential energy surface.
lsqfit_op_0*.{inp,out}Inputs and outputs for the parameter least-squares fitting using the original multiplicity along with fits using alternate multiplicities (
grep "Final RMSE" *.outto see RMSE for all runs).mm*.prmFinal parameters from the fits with the different multiplicities.
*.datFiles containing dihedral angles and energies for fitting. Note that the
mm_init_ener_0.datis the MM PES with the targeted dihedral parameters set to zero.
Practical considerations¶
The use of the CGenFF penalty scores to select dihedrals for optimization in conjunction with the selection of only rotatable bonds is designed for an automated approach to dihedral parameter optimization. However, this may often lead to multiple dihedrals being selected for optimization, which is computationally demanding, or no dihedrals being selected based on low or zero penalty scores, where zero indicates that the specific parameter is already in the CGenFF force field and, by default, such dihedrals will not be listed as available for optimization. However, in cases where the user is concerned about the accuracy of a particular rotatable bond even when it is already in the CGenFF parameter set (e.g., a rotatable bond involved in the link between two ring systems that are part of a lead compound), then the user may specifically identify the atom types defining that dihedral parameter(s) and input them individually. While the penalty scores represent a useful metric by which to judge the quality of a dihedral parameter it should be noted that the CGenFF penalty scores are based on analogy to known parameters. Accordingly, a parameter with a high penalty may yield acceptable conformational energies, while a parameter already in CGenFF (i.e., penalty = 0) may not yield an acceptable energy surface due to, for example, the terminal rings about the associated bond creating a significantly different context than that in which the dihedral parameter was initially optimized.
An important consideration is molecular size. As the fitting procedure requires QM calculations the size of the molecule under study significantly impacts the speed of the overall fitting procedure. To limit the computational demand while achieving the desired accuracy in the parameters it is suggested that compounds be subdivided into model compounds extracted from that full compound that contain two terminal ring systems along with the linker between those rings. The number of substituents on each ring should be limited to those deemed essential to represent the chemical character of the rings while minimizing the number of substituents to limit computational costs. For example a compound with three ring systems with two linkers would be subdivied into two model compounds with two rings each, with one of the rings common to both model compounds. Once the two individual CGenFF-optimizer runs are finished, the new parameters may be combined and added to your the CGenFF parameter files.
During parameter fitting, the program initially uses the dihedral parameter
multiplicities assigned by the CGenFF program. Once the RMSE is returned, the
program then does additional fitting with a multiplicity of 1, then
multiplicities of 1 and 2, 1, 2 and 3 and 1, 2, 3 and 6. The current
implementation outputs the results from all the multiplicities tested. As the
lowest RMSE will generally be with multiplicities 1,2,3,6 the user may select a
set of parameters with different multiplicities. Those parameters are in the
files raw_data/mm*.prm. Note that the multiplicities of 4 and 5 are omitted as
these are rarely used for rotatable bonds even though, in same cases, the CGenFF
program will assign dihedral parameters with these multiplicities and the
program will initially include a dihedral with a multiplicity of 4 or 5 in the
initial fitting.
If the level of agreement using the final parameters is not satisfactory it
suggests hysteresis in the energy surface. To overcome this the user may
consider focusing the fitting on an ‘appropriate’ part of the energy surface by
trimming down some data-points on the QM & MM energies to get a better fit to
the low energy region of the energy surface. To achieve this consider running
${SILCSBIODIR}/cgenff-optimizer/refit_par_with_subsurface
dih_qm_<dihno>.dat dih_mm_<dihno>.dat (Under development).
Upon completion of the fitting a subdirectory ‘raw_data’ is created that contains an extensive number of data file with the various QM energies, MM energies and additional information. In addition, the subdirectory ‘raw_data/psi4_calc’ contains the inputs and outputs from the psi4 QM optimizations. These files may be used for additional fitting or analysis.
In cases where the least-squares fitting is not completed successfully, possibly
due to all the QM jobs not finishing, fitting alone may be performed if all the
QM data is directly available in subdirectory psi4_calc (not
raw_data/psi4_calc). For example, if a subset of the QM jobs did not
finished the job input scripts in psi4_calc (e.g., psi4_inp_xxxx_1_23.inp)
maybe submitted directly to complete those jobs followed by resubmission of the
cgenff-optimizer command from the parent directory.