SILCS: Site Identification by Ligand Competitive Saturation¶
SILCS simulation background¶
The design of small molecules that bind with optimal specificity and affinity to their biological targets, typically proteins, is based on the idea of complementarity between the functional groups in a small molecule and the binding site of the target. Traditional approaches to ligand identification and optimization often employ the one binding site/one ligand approach. While such an approach might be straightforward to implement, it is limited by the resource-intensive nature of screening and evaluating the affinity of large numbers of diverse molecules.
Functional group mapping approaches have emerged as an alternative, in which a series of maps for different classes of functional groups encompass the target surface to define the binding requirements of the target. Using these maps, medicinal chemists can focus their efforts on designing small molecules that best match the maps.
Site Identification by Ligand Competitive Saturation (SILCS) offers rigorous free energy evaluation of functional group affinity pattern for the entire 3D space in and around a protein [7]. The SILCS method yields functional group free energy maps, or FragMaps, which are precomputed and then used to rapidly facilitate ligand design. FragMaps are generated by molecular dynamics (MD) simulations that include protein flexibility and explicit solvent/solute representation, thus providing an accurate, detailed, and comprehensive set of data that can be used in database screening, fragment-based drug design, and lead optimization of small molecules.
In the context of biological therapeutics, the comprehensive nature of the FragMaps is of utility for excipient design as all possible binding sites of all possible excipients and buffers can be identified and quantified.
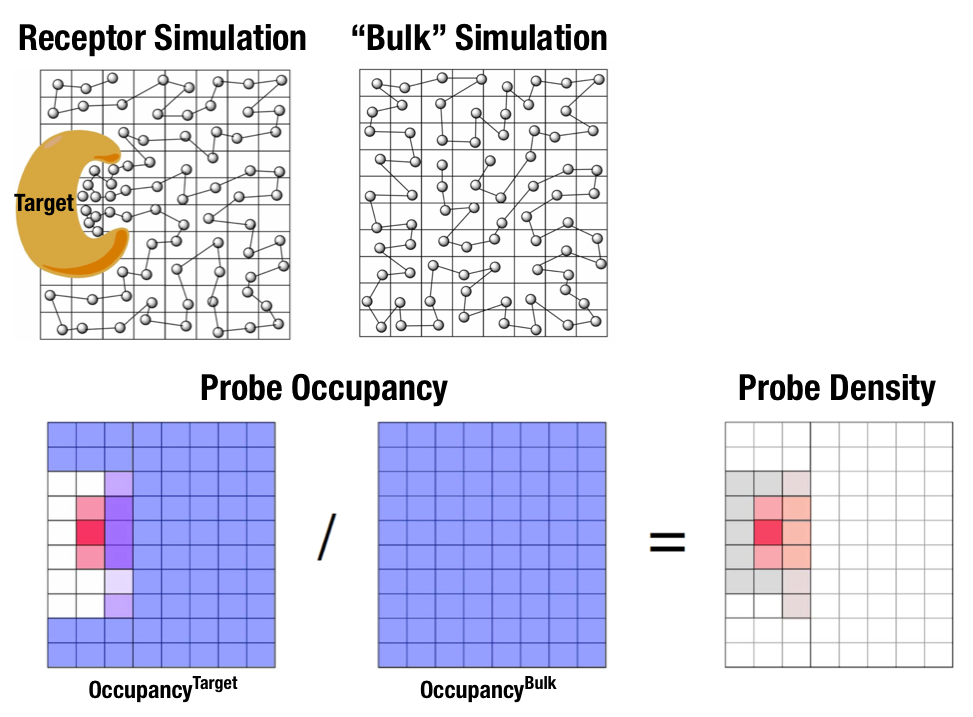
Fig. 1 Illustration of fragment density maps.¶
Fig. 1 illustrates FragMaps generation. Two MD simulations of a probe molecule (e.g., benzene) are performed in the presence of a protein and in aqueous solution (without protein), resulting in two occupancy maps, \(O^\mathrm{target}\) and \(O^\mathrm{bulk}\), respectively. The GFE (grid free energy) FragMaps are then derived by the following formula
By generating FragMaps using various small solutes (probes) that include a diversity of atoms and chemical functional groups, it is possible to map the functional group affinity pattern of a protein. FragMaps encompass the entire protein such that all FragMap types are in all regions. Thus, information on the affinity of all the different types of functional groups is available in and around the full 3D space of the protein or other target molecule.
A Standard SILCS simulation uses benzene, propane, methanol, imidazole, formamide, dimethylether, methylammonium, acetate, and water as probes to generate Standard SILCS FragMaps. A Halogen SILCS simulation uses fluorobenzene, chlorobenzene, bromobenzene, fluoroethane, chloroethane, and trifluoroethane as probes to generate Halogen SILCS FragMaps. Halogen SILCS FragMaps are used to augment the information provided by Standard SILCS FragMaps.
FragMaps may be used in a qualitative fashion to facilitate ligand design by allowing the medicinal chemist to readily visualize regions where the ligand can be modified or functional groups added to improve affinity and specificity. As the FragMaps include protein flexibility, they indicate regions of the target protein that can “open” thereby identifying regions under the protein surface accessible for ligand binding. In addition to FragMaps, an Exclusion Map is generated based on regions of the system that are not sampled at all by any probe molecules, including water, during the MD simulations. Therefore, the Exclusion Map represents a strictly inaccessible surface.
Many proteins contain binding sites that are partially or totally inaccessible to the surrounding solvent environment that may require partial unfolding of the protein for ligand binding to occur. SILCS sampling of such deep or inaccessible pockets is facilitated by the use of a Grand Canonical Monte Carlo (GCMC) sampling technique in conjunction with MD simulations [10]. Thus, the SILCS technology is especially well-suited for targeting the deep or seemingly inaccessible binding sites found in targets like GPCRs and nuclear receptors.
Once a set of ligand-independent SILCS FragMaps are produced, they can be used for various purposes that rapidly rank ligand binding in a highly computationally efficient manner for multiple ligands WITHOUT recalculating the FragMaps. Applications of SILCS methodology include:
Binding site identification
Binding pocket searching with known ligands
Binding pocket identification via pharmacophore generation
Binding pocket identification via fragment screening
Database screening
SILCS 3D pharmacophore models
Ligand posing using available techniques (Catalyst, etc)
Ligand ranking
Fragment-based ligand design
Identification of fragment binding sites
Estimation of ligand affinity following fragment linking
Expansion of fragment types
Ligand optimization
Qualitative ligand optimization by FragMaps visualization
Quantitative evaluation of ligand atom contributions to binding
Quantitative estimation of relative ligand affinities
Quantitative estimation of large numbers of ligand chemical transformations
SILCS generates 3D maps of interaction patterns of functional group with your target molecule, called FragMaps. This unique approach simultaneously uses multiple different small solutes with various functional groups in explicit solvent MD simulations that include target flexibility to yield 3D FragMaps that encompass the delicate balance of target-functional group interactions, target desolvation, functional group desolvation and target flexibility. The FragMaps may then be used to both qualitatively direct ligand design and quantitatively to rapidly evaluate the relative affinities of large numbers of ligands following the precomputation of the FragMaps.
This chapter goes over the workflow for generating SILCS FragMaps.
FragMap names and underlying probe atoms¶
Each FragMap generated from a SILCS simulation is the result of computing occupancies for one or more kinds of atoms associated with the SILCS probes. The tables below provide the complete lists for Standard SILCS and for Halogen SILCS simulations.
FragMap name |
generated from probe atom(s) |
|---|---|
acec |
acetate carboxylate carbon |
apolar |
generic apolar carbons (benc+prpc) |
benc |
benzene carbons |
dmeo |
dimethyl ether oxygen |
forn |
formamide nitrogen |
foro |
formamide oxygen |
hbacc |
generic hydrogen bond acceptors (foro+dmeo+imin) |
hbdon |
generic hydrogen bond donors (forn+iminh) |
imin |
imidizole nitrogen (h-bond acceptor) |
iminh |
imidizole nitrogen (h-bond donor) |
mamn |
methylamine nitrogen |
meoo |
methanol oxygen |
prpc |
propane carbon |
tipo |
water oxygen |
FragMap name |
generated from probe atom |
|---|---|
brbx |
bromobenzene bromine |
clbx |
chlorobenzene chlorine |
clex |
chloroethane chlorine |
fetx |
fluoroethane fluorine |
flbc |
fluorbenzene fluorine |
tfec |
trifluoroethane carbon bonded to fluorines |
The FragMap with the name “excl” is a special case. It is the SILCS Exclusion Map, and it enumerates all voxels where no probe molecule (including water) sampling occurred.
Running SILCS simulations from the SilcsBio GUI¶
Please see SILCS simulation from the GUI as described in Graphical User Interface Quickstart for a step-by-step description for using the SilcsBio GUI for SILCS simulations.
FragMap generation using SILCS begins with a well-curated protein structure file (without ligand) in PDB file format. We recommend keeping only those protein chains that are necessary for the simulation, removing all unnecessary ligands, renaming non-standard residues, filling in missing atomic positions, and, if desired, modeling in missing loops. Please contact support@silcsbio.com if you need assistance with protein preparation.
A typical SILCS simulation can produce output totaling in excess of 100 GB, so please select a “Project Directory” folder on your server with appropriate free space. The default is determined by the “Project Directory” setting during Remote server setup.
The setup process automatically builds the topology of the simulation system, creating metal-protein bonds if metal ions are present, rotates side chain orientations to enhance sampling, and places probe molecules around the protein. If missing loops were not modeled into the input PDB file, the amino acids on either side of each missing loop will be automatically capped with neutral functional groups.
During setup, you will need to decide whether to run a Standard SILCS Simulation, Standard + Halogen SILCS Simulations, or a Halogen SILCS Simulation (with another project’s Standard SILCS FragMaps). All functionality in the SILCS platform requires FragMaps from a Standard SILCS Simulation. FragMaps from Halogen SILCS Simulations can supplement Standard SILCS FragMaps for silcs/silcs-mc-optimization and for silcs/silcs-mc-docking. Visualization of Halogen SILCS FragMaps can be very useful for qualitatively informing ligand design (see silcs/visualization). See FragMap names and underlying probe atoms for a complete listing of both Standard and Halogens SILCS probe molecules and the resulting FragMaps.
Running a Halogen SILCS simulation requires the same omputation time as running a Standard SILCS simulation, so selecting “Standard + Halogen SILCS Simulations” will consume double the computational resources as selecting “Standard SILCS Simulation”. The default is to generate and run 10 systems each for a Standard SILCS simulation and for a Halogen SILCS simulation. Therefore, if your computing resource has 10 nodes, it will take twice as much wall time to complete a “Standard + Halogen SILCS Simulations” job as to complete a “Standard SILCS Simulation”. However, if you have access to 20 nodes simultaneously, the wall time will be the same, since each individual system is run independently of the others.
While the same identical input protein coordinates are used for constructing each of these systems, each system is unique because the probe molecules are randomly placed. Additional diversity is added by default to initial coordinates by scrambling the sidechains of exposed amino acids.
Tip
Advanced Settings options during setup allow you to change the scrambling of sidechains (default: on), the number of systems (default: 10), and the simulation temperature (default 298 K). We STRONGLY urge you to use these default settings, which have been selected based on extensive experience.
It is also possible to choose an existing setup folder or an existing trajectory folder at this time.
Once the systems are set up, you will need to start the SILCS simulations. There are separate tabs for running the Standard SILCS simulation and the Halogen SILCS simulation, and you will need to use each tab and the “Run…” button underneath that tab to run the corresponding simulation type.
Tip
Advanced Settings options allow you to change the number of systems that are run, as well as the number of processors used for each run and the length of each run. Leaving these fields blank will result in default values being used. We STRONGLY urge you to use the default settings. If you have concerns about the number of processors per run, please contact your system administrator.
Tip
If a simulation stops prematurely, the progress bar will turn red and show a “Restart” option. When the job is restarted, the simulation will continue from the last cycle, but the progress bar may reset. This is because job recovery iterates over existing cycles to check if output was correctly produced. The completed cycles will be automatically skipped and the progress will update to where the job had stopped. That is to say, restarting a failed or stopped job will not cause you to lose existing job progress.
Running SILCS simulations from the command line interface¶
Please see SILCS simulation from the command line as described in Command Line Interface Quickstart for a step-by-step description for using the Command Line Interface (CLI) for SILCS simulations.
FragMap generation using SILCS begins with a well-curated protein structure file (without ligand) in PDB file format. We recommend keeping only those protein chains that are necessary for the simulation, removing all unnecessary ligands, renaming non-standard residues, filling in missing atomic positions, and, if desired, modeling in missing loops. Please contact support@silcsbio.com if you need assistance with protein preparation.
SILCS simulation setup¶
${SILCSBIODIR}/silcs/1_setup_silcs_boxes prot=<Protein PDB>
Warning
The setup program internally uses the GROMACS utility pdb2gmx, which may
have problems processing the protein PDB file. The most common reasons for
errors at this stage involve mismatches between the expected residue
name/atom names in the input PDB and those defined in the CHARMM force-field.
To fix this problem: Run the pdb2gmx command manually from within the
1_setup directory for a detailed error message. The setup script already
prints out the exact commands you need to run to see this message.
cd 1_setup
pdb2gmx –f <PROT PDB NAME>_4pdb2gmx.pdb –ff charmm36 –water tip3p –o test.pdb –p test.top -ter -merge all
Once all the errors in the input PDB are corrected, rerun the setup script.
The setup command offers a number of options. The full list of options can be reviewed by simply entering the command without any options:
$SILCSBIODIR/silcs/1_setup_silcs_boxes
This script will setup Topology/PDB for SILCS simulations
Usage: $SILCSBIODIR/silcs/1_setup_silcs_boxes prot=<protein PDB file>
Optional Parameters:
numsys=<# of simulations; default=10>
skip_pdb2gmx=<true/false; default=false>
lig=<lig.mol2; default=empty/NULL>
core_restraints_string=<restrained residues, e.g., "r 17-21 | r 168-174"; default=all>
scramblesc=<true/false; default=true>
margin=<system margin in Angstrom; default=15>
halogen=<true/false; default=false>
core_restraints_string: By default, a weak harmonic positional restraint, with a force constant of ~0.1 kcal/mol/Å2, is applied to all C-alpha atoms during SILCS simulations. This option lets you modify this default.
For example, if
core_restraints_string="r 17-160 | r 168-174"is supplied, then only the C-alpha atoms in residues 17 to 160 and residues 168 to 174 will be restrained.lig: Use this option if your input structure contains a cofactor and you wish to compute your FragMaps in the presence of that cofactor. Remove the cofactor from the input PDB, and provide the structure of the cofactor in
.mol2format using this option. The cofactor needs to be aligned appropriately with the protein structure.
Note
If your input structure contains a metal ion, please see What happens when I set up SILCS simulations with an input structure containing a metal ion?
SILCS GCMC/MD simulation¶
Following completion of the setup, run 10 GCMC/MD jobs using the following script:
${SILCSBIODIR}/silcs/2a_run_gcmd prot=<Protein PDB>
This script will submit 10 jobs to the predefined queue. Each job runs out 100 ns of GCMC/MD with the protein and the solute molecules.
Tip
SILCS simulations are run at a temperature of 298 K by default, and most
published work with SILCS has used this default temperature. It is
possible to set a custom temperature by adding the
temp=<simulation temperature; default=298> option to the
2a_run_gcmd command. For example, temp=310 would run the SILCS
simulations at 310 K, which may enhance protein conformational sampling.
Warning
It is important to pass the same temp= option to 2c_fragmap (see
below) as was used for 2a_run_gcmd. The simulation temperature is
required for Boltzmann conversion of occuppany FragMaps to Grid Free Energy
(GFE) FragMaps, and needs to be the same across both commands for
correctness. The temp= option can be safely ommitted from both of these
steps if the default temperature of 298 K is desired.
Once the simulations are complete, the 2a_run_gcmd/[1-10] directories will
contain *.prod.125.rec.xtc trajectory files. If these files are not
generated, then your simulations are either still running or stopped due to a
problem. Look in the log files within these directories to diagnose problems.
FragMap generation¶
Once your simulations are done, generate the FragMaps using the following command:
${SILCSBIODIR}/silcs/2b_gen_maps prot=<Protein PDB>
This will submit 10 single-core jobs that will build occupancy maps (FragMaps) spanning the simulation box for select probe molecule atoms representing different functional groups. For a ~50K atom simulation system, this step takes approximately 10-20 minutes to complete. The FragMaps have a default grid spacing of 1 Å.
The next step is to combine the occupancy FragMaps generated from individual simulations and convert them into GFE FragMaps. Each voxel in a GFE FragMap contains a Grid Free Energy (GFE) value for the probe type that was used to create the corresponding occupancy FragMap.
${SILCSBIODIR}/silcs/2c_fragmap prot=<Protein PDB>
Warning
It is important to pass the same temp= option to 2c_fragmap
as was used for 2a_run_gcmd (see above). The simulation temperature is
required for Boltzmann conversion of occuppany FragMaps to Grid Free Energy
(GFE) FragMaps, and needs to be the same across both commands for
correctness. The temp= option can be safely ommitted from both of these
steps if the default temperature of 298 K is desired.
GFE FragMaps will be created in the silcs_fragmap_<protein PDB>/maps
directory, and PyMOL and VMD scripts to load these file will be created in the
silcs_fragmap_<protein PDB> directory.
Cleanup¶
Raw trajectory output files are large. To reduce disk usage due to these files, use the cleanup command:
${SILCSBIODIR}/silcs/2d_cleanup prot=<Protein PDB>
The 2d_cleanup command deletes unnecessary files, such as those from the
equilibration phase, and deletes hydrogen atoms from production trajectory
files for each run directory, 1/, 2/, etc. It then creates a
.tar.gz file of that directory. That is to say, the contents of
1.tar.gz and 1/ after running cleanup are identical. You may choose to
remove either one of those without loss of information, though we strongly
recommend comparing the output of tar tvzf <run>.tar.gz with the output of
find <run> -type f, where you replace <run> with 1, 2, etc. and
confirm the contents are identical before deleting anything.
SILCS simulation setup with GPCR targets¶
You may use either a bare protein structure or your own pre-built protein/bilayer system with SILCS.
If you are beginning with a bare protein, SilcsBio provides a command line utility to embed transmembrane proteins, such as GPCRs, in a bilayer of 9:1 POPC/cholesterol for subsequent SILCS simulations:
${SILCSBIODIR}/silcs-memb/1a_fit_protein_in_bilayer prot=<Protein PDB>
This command will align the first principal axis of the protein with the bilayer normal (Z-axis) and translate the protein center of the mass to the center of the bilayer (Z=0).
If the protein is already oriented as desired, orient_principal_axis=false
will suppress automatic principal axis-based alignment.
${SILCSBIODIR}/silcs-memb/1a_fit_protein_in_bilayer prot=<Protein PDB> orient_principal_axis=false
By default the system size is set to 120 Å along the X and Y dimensions. To
change the system size, use the bilayer_x_size and bilayer_y_size
options.
${SILCSBIODIR}/silcs-memb/1a_fit_protein_in_bilayer prot=<Protein PDB> bilayer_x_size=<X dimension> bilayer_y_size=<Y dimension>
The resulting protein/bilayer system will be output in a PDB file with suffix
_popc_chol. It is important to use molecular visualization software at
this stage to confirm correct orientation of the protein and size of the bilayer
before using this output as the input in the next step.
Alternatively, you may use a protein/bilayer system that has been built using other software tools.
The following command will prepare the SILCS simulation box by adding water and probe molecules to the protein/bilayer system:
${SILCSBIODIR}/silcs-memb/1b_setup_silcs_with_prot_bilayer prot=<Protein/bilayer PDB>
SILCS GCMC/MD simulation can now be launched with the following command:
${SILCSBIODIR}/silcs/2a_run_gcmd prot=<Protein/bilayer PDB> memb=true
The SILCS GCMC/MD will begin with a 6-step pre-equilibration to slowly relax the bilayer followed by 10 ns of relaxation of the protein into the bilayer prior to the production simulation.
Tip
GPCR simulation systems are typically large and can require significant storage space (> 200 GB) and simulation time (> 14 days with GPUs).
FragMap generation from the GCMC/MD simulation data follows the same protocol as
for non-bilayer systems. Refer to previous instructions for
$SILCSBIODIR/silcs/2b_gen_maps and $SILCSBIODIR/silcs/2c_fragmap.
If you encounter a problem, please contact support@silcsbio.com.
SILCS simulation setup with halogen probes¶
SILCS probes with halogen-containing functional groups can be useful for extending the diversity of chemical space covered by FragMaps. While a common approach is to consider halogen atoms as non-polar groups, research on non-covalent “halogen bonding” suggests halogen atoms warrant special treatment.
The CHARMM General Force Field CGenFF v.2.3.0+ allows halogen atoms to be treated with lonepairs to achieve directionality in polar interactions. These force field improvements are included in SILCS simulations from version 2020.1 of the SilcsBio software. Halogen-protein interactions are determined using a set of halogenated probes in SILCS simulations: chlorobenzene, fluorobenzene, bromobenzene, chloroethane, fluoroethane, and trifluoroethane.
To use these halogenated SILCS probes in your simulation instead of the
standard SILCS probes, add the halogen=true keyword:
${SILCSBIODIR}/silcs/1_setup_silcs_boxes prot=<Protein PDB> halogen=true
${SILCSBIODIR}/silcs/2a_run_gcmd prot=<Protein PDB> halogen=true
${SILCSBIODIR}/silcs/2b_gen_maps prot=<Protein PDB> halogen=true
${SILCSBIODIR}/silcs/2c_fragmaps prot=<Protein PDB> halogen=true
This will create folders ending with _x (i.e., 1_setup_x/, etc.)
to denote SILCS-Halogen simulation systems, as opposed to standard SILCS
simulations. The resulting halogen FragMaps will be included in the
silcs_fragmaps_<PROT>/maps folder. To use halogen FragMaps in
SILCS-MC jobs, refer to the halogen=true option in
SILCS-MC: Docking and Pose Refinement.
While running SILCS with halogen probes is currently supported only through the command line interface, the SilcsBio GUI will automatically load halogen FragMaps from these simulations alongside standard FragMaps.
If you encounter a problem, please contact support@silcsbio.com.
Resuming stopped SILCS jobs¶
Situations such as exceeding workload manager (job queue) walltime limits can
cause SILCS jobs to stop before running to completion. Resuming such jobs from
where they stopped is straightforward. On the server where the job was running,
go to the directory <Project Location>/2a_run_gcmd/i, where i is an
integer from 1 to 10 and corresponds to the stopped job among the ten SILCS
GCMC/MD jobs that were being run for that particular target. In that directory,
directly use the file job_mc_md.cmd to submit the job to the workload
manager, for example qsub job_mc_md.cmd for Sun Grid Engine (SGE) and
sbatch job_mc_md.cmd for Slurm.
To ensure a successful restart, make sure to issue any required extra setup
commands (for example, as listed in the “Extra setup” field under
from the SilcsBio GUI Home screen) such as
export LD_LIBRARY_PATH=<path/to/cuda/libraries> or
module load <name of cuda module> before submitting the job.