SILCS-Covalent
Background
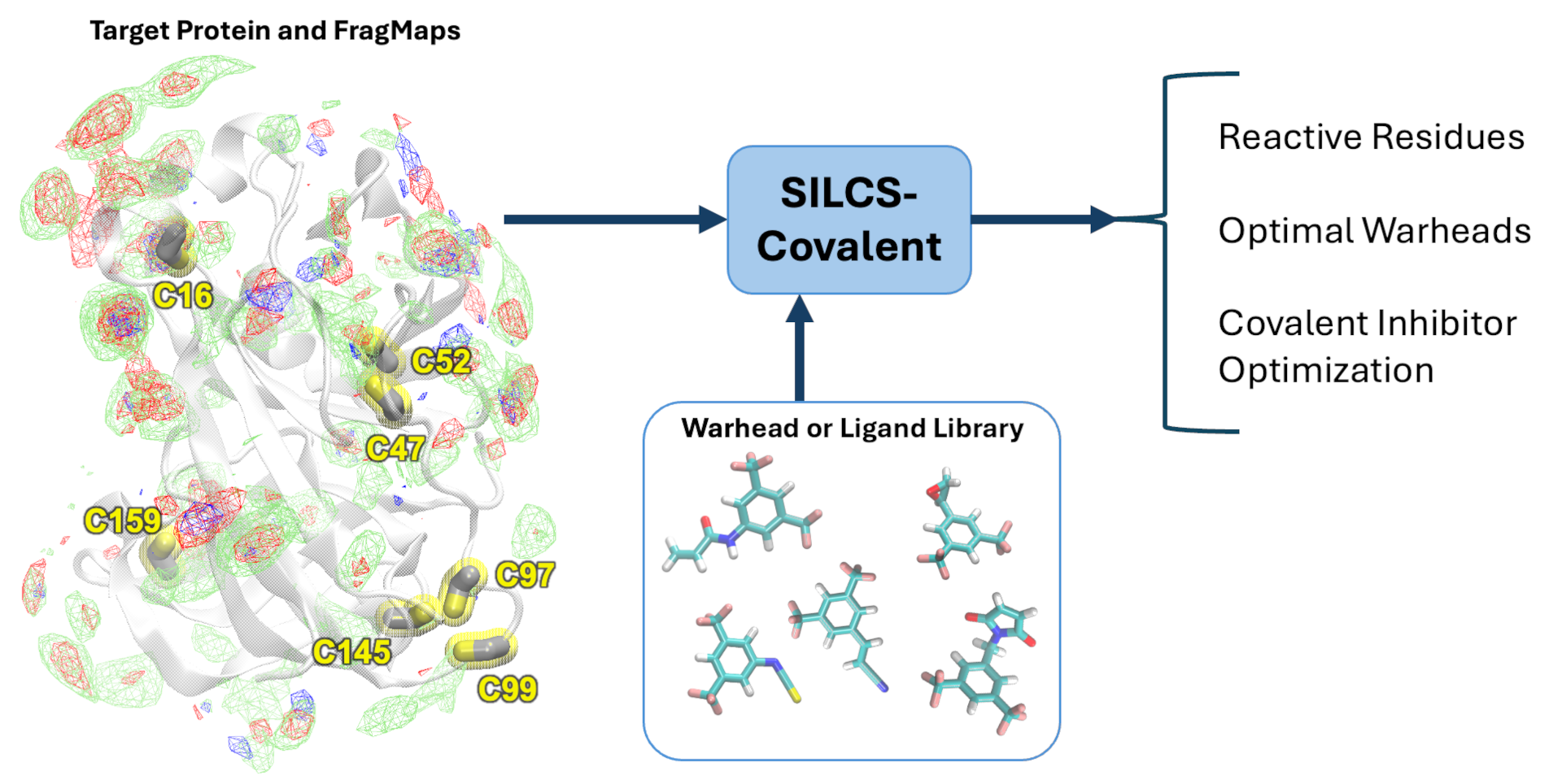
The SILCS-Covalent workflow is a comprehensive approach to covalent drug design that combines molecular simulations, fragment-based binding site identification, and machine learning models. This workflow addresses the challenges of designing covalent drugs by providing insights into the binding preferences and reactivity of potential covalent ligands. It utilizes SILCS FragMaps to identify reactive cysteine residues and select optimal warhead groups. Other covalent linkages are also supported. Machine learning models trained on activity data further aid in predicting the efficacy and selectivity of warheads. The workflow provides the efficiency and reliability in identifying potential covalent ligands and optimizing their non-covalent interactions. Additional details on SILCS-Covalent are available in reference [21].
Note
The SILCS-Covalent workflow is slightly different from the SILCS-MC Docking in Restrained Mode workflow. The SILCS-MC Docking in Restrained Mode workflow is designed to run the SILCS-MC Docking protocol with harmonic restraint on selected atoms of the ligand. The SILCS-Covalent workflow is designed to run the SILCS-MC docking without any restraints on the reactive atom of the ligand first and then run the analysis to identify the ligand conformations with reactive atoms located within a certain distance from the reactive atoms of the residues in the binding site. The SILCS-Covalent workflow allows the reactivity of the target residues and the warheads or ligands to be assessed, whereas the SILCS-MC Docking in Restrained Mode workflow only identifies energetically favored binding poses given that a covalent bond is formed between a target residue and the ligand.
SILCS-Covalent Using the SilcsBio CLI
Launch SILCS-Covalent docking runs:
To set up and run SILCS-Covalent docking from the command line interface, create a directory containing all the ligands to be evaluated. Each ligand can be stored as a separate SDF or Mol2 file. Alternatively, all the ligands can be combined into a single SDF file. With this information, enter the following command to set up and launch SILCS-Covalent docking runs:
${SILCSBIODIR}/silcs-covalent/1_run_silcs_covalent prot=<prot pdb>Required parameters:
Path and name of protein PDB file:
prot=<protein pdb file>Optional parameters:
Mode of the SILCS-Covalent workflow:
mode=< warhead or ligand >The mode of the SILCS-Covalent worflow can be set to either
warheadorligand. The default mode iswarhead.
In
ligandmode, the workflow will only run the SILCS-MC Docking protocol on the ligands provided to identify the most favorable binding poses of the ligands in the binding site around the linking residue(s). In theligandmode, the user must provide the ligands in SD file or Mol2 format using either theligdirorsdfileoption.In
warheadmode, the workflow will run the SILCS-MC Docking protocol on the warheads provided to identify the most favorable binding poses of the warheads in the binding site around the linking residue.Additionally in
warheadmode, the user may provide additional information on the experimental data of the warheads such as kGSH, the glutathione (GSH) reactivity rate constant. The experimental data can be used to train machine learning models to quantify the effectiveness of various warhead groups for covalent drug design for the given linking residue of your target protein. Currently, this feature is only available for the cysteine (CYS) residues as the linking residue and an ML model trained on the Petri’s Warhead Library [22] will be used to predict the activity class for the warheads specific to the linking cysteine residue.Please refer to reference [21] for more details.
Path and name of directory containing ligand SD or Mol2 files:
ligdir=<ligand directory>If the
ligdiroption is used, only one molecule per file underligdirwill be processed for SILCS-MC docking. For an SD/SDF file containing multiple molecules, usesdfile=<path to sdfile>instead of theligdiroption.Path and name of output directory for the SILCS-Covalent workflow:
covaldir=<output directory default=4_covalent_warhead>The residue name of the amino acid for covalent link with the ligand:
linkresname=<residue name for covalent link; CYS/LYS/SER/THR; default=CYS>Currently, only cysteine (CYS), lysine (LYS), serine (SER), and threoine (THR) residues are supported. Asparagine (ASN) residues will be supported in the future.
The residue ID of the bound amino acid for covalent bonding with the ligand:
linkresid=<residue ID for covalent link; default=NULL>By default, SILCS-MC docking of each warhead or ligand will be performed in proximity to each residue with residue names specified by
linkresnameor each cysteine of the target protein if thelinkresnameoption is not used. If thelinkresidoption is specified, then the SILCS-MC docking will only be performed in proximity to the residue with the residue ID specified bylinkresidwill be targeted.The residue ID(s) of amino acids disqualified from covalent bonding with the ligand:
skipresid=<residue IDs to skip; e.g. "145,231"; default=NULL>Certain residues may be ommitted from consideration in the SILCS-MC docking by specifying their residue IDs with the
skipresidoption. This option can be useful in omitting docking calculations near cysteine residues involved in disulfide bonds.Path and name of directory containing FragMaps:
mapsdir=<location and name of directory containing FragMaps; default=maps>The supplied
mapsdirshould contain the protein side chain probability maps. The probability maps for each type of residue are generated by default during thesilcs/2b_gen_mapsandsilcs/2c_fragmapsteps in the latest version of the SilcsBio software. Probability maps for a specific residue ID can be generated by re-running thesilcs/2b_gen_mapsandsilcs/2c_fragmapsteps with theresidmapoption. Please refer to SILCS Simulations Using the CLI for more details.Use GPU for the SILCS-MC Docking protocol:
gpu=<true/false; default=true>When
gpu=falseand if thebundleoption is set to true, the workflow will run the SILCS-MC Docking protocol in parallel using multiple processors. The number of processors can be specified using thenprocoption. The default value fornprocis the number of processors available on the system.When
gpu=true, thebundle=trueoption works differently. Thebundleoption will bundlenprocnumber of SILCS-MC Docking runs into a single job. The default value fornprocis the number of processors available on the system.bundle=<true/false; default=false> nproc=<# of processor used when bundle=true>Translation distance for defining sampling spheres in six directions around the reactive atom of the linking residue in Å:
translation=<translation distance for defining sampling spheres; default=3.5>Path and name of the custom parameters file for the SILCS-MC Docking protocol:
paramsfile=<custom params file>By default, the SILCS-MC Docking protocol in the SILCS-Covalent workflow uses the parameters file located in
${SILCSBIODIR}/templates/silcs-covalent/params-<warhead/ligand>.tmpl.
Evaluate docked poses:
After the SILCS-Covalent docking runs are completed, the docked poses are evaluated to identify the most favorable binding poses of the ligands in the binding site around the linking residue.
To evaluate the docked poses, enter the following command:
${SILCSBIODIR}/silcs-covalent/2_calc_lgfe_probres prot=<prot pdb> covaldir=<name of output directory>The above command will evaluate the poses based on three criteria:
eq: Best pose when sorted by LGFE score
rx: Best pose when sorted by ProbRes score with LGFE < 0 kcal/mol
rxeq: Best pose when sorted by LGFE score with a ProbRes score greater than the ProbRes cutoff value. This pose will have both a favorable LGFE score and potential for covalent bond formation.Required parameters:
Path and name of protein PDB file:
prot=<protein pdb file>Path and name of output directory for the SILCS-Covalent workflow:
covaldir=<output directory default=4_covalent_warhead>Optional parameters:
Path and name of the file containing the list of warhead/ligand name and reactive atom IDs:
whidfile=<file with id list for warhead/ligand reactive atom; default="Petri's Warhead Lib">The file specified by the
whidfileoption should contain two columns. In the first column, the warhead or ligand names are listed. In the second column, the reactive atom IDs of the warheads or ligands are listed:Each line should have two columns:
<warhead_name/ligand_name> <reactive_atom ID>By default, the
whidfilefor Petri’s Warhead Library is used. The Petri’s Warhead Librarywhidfilecan be found in${SILCSBIODIR}/data/databases/petri_warhead.whid.Distance cutoff for covalent bond definition in Å:
dcutoff=<distance cutoff for covalent bond definition; default=2.0>The reactive atoms of the ligand and linking residue for a given docked pose are considered to be sufficiently close to form a covalent bond if the distance between the two atoms is within the cutoff distance defined by the
dcutoffoption.ProbRes cutoff for filtereing poses unlikely to form covalent bonds:
prcutoff=<cutoff value for ProbRes; default=0.025>The probability of the reactive atoms of the ligand and linking residue for a given docked pose to be within the cutoff distance defined by
dcutoffis represented by ProbRes. Poses that do not have ProbRes larger than the cutoff defined by theprcutoffare filtered from further analysis.Number of output poses:
npose=<number of best scoring poses; default=1>By default, only three the best scoring, based on LGFE (
eq), ProbRes (rx), and PRCutoff (rxeq), docked poses will be output. To output additional docked poses, use thenposeoption and specify the desired number of docked poses. E.g., if 5 poses are desired, usingnpose=5will result in the 5 best scoring poses being output for each warhead or ligand and each basis.Option to output docked poses in PDB format:
pdb=<true/false; default=false>When
pdb=truethe docked poses will be output in PDB format in addition to the default SD file format.Option to calculate 4DBA descriptors, and 4DBA and LGFE-4DBA scores:
fourdba=<true/false; default=false>When
fourdba=truethe four 4DBA descriptors of each input ligand will be calculated and output intolgfe-4dba.csv. A high 4DBA score indicates that the ligand is bioavailable while a low 4DBA score indicates that the ligand is not bioavailable. For more information of 4DBA and LGFE-4DBA scores, please see 4D Bioavailability (4DBA) Calculation. Therdkit,tqdm,ipythonandscipypackages must be installed for the 4DBA score calculation. Please refer to Python 3 Requirement for more information.Submit job to queuing system:
batch=<submit job to queuing system; true/false; default=false>Path to the Python executable:
python=<path to python executable; default=${python}>The default python path can be checked using the
which pythoncommand.
Assessment of SILCS-Covalent Results in CLI
The silcs-covalent/2_calc_lgfe_probres command will generate a number of output
files that can be used to identify the residues most ammenable to covalent bonding,
identify the optimal warheads or ligands for a target protein or specific residues
within a target protein, or optimize covalent inhibitors.
In both warhead and ligand modes, three poses in SD file format are output
into <covaldir>/<reactive residue>/minconfpdb/ for each warhead or ligand, where
<reactive residue> is the linking residue in proximity to which the warhead or
ligand was docked. The three poses correspond to the most favorable poses based on
LGFE (eq), ProbRes (rx), and PRCutoff (rxeq):
<ligand|warhead>.eq.sdf: Pose with the most favorable energy (lowest LGFE).
<ligand|warhead>.rx.sdf: Pose with the highest probability of ligand and linking residue reactive atoms being in proximity (highest ProbRes) with favorable energy (LGFE < 0 kcal/mol).
<ligand|warhead>.rxeq.sdf: Pose with the most favorable energy (lowest LGFE) with the probability of the ligand and linking residue reactive atoms being in proximity greater than a cutoff value (ProbRes > 0.025 by default). This pose will have both a favorable LGFE score and potential for forming a covalent bond with the reactive residue.
In addtion, for each reactive residue in proximity to which the warheads or ligands
were docked, a summary of the LGFE and ProbRes of each of the three warhead or ligand
poses (eq, rx, rxeq) will be output in
<covaldir>/<reactive residue>.<ligand|warhead>.csv. Below are the contents of
each column of <covaldir>/<reactive residue>.<ligand|warhead>.csv:
In the first column, the warhead or ligand names are listed.
In the second, third, and fourth columns, the LGFEs of the most favorable poses of the corresponding warhead or ligand in terms of LGFE (
eq), ProbRes (rx), and PRCutoff (rxeq), respectively, are listed.In the fifth, sixth, and seventh columns, the ProbRes values of the most favorable poses of the corresponding warhead or ligand in terms of LGFE (
eq), ProbRes (rx), and PRCutoff (rxeq), respectively, are listed.
In warhead mode with cysteine residues specified as the linking residue
and the Petri’s Warhead Library [22] specified as the warheads,
in addition to the output results described above, predicted
kGSH, the GSH reactivity rate constant, and activity classifications are
output in <reactive cysteine>.rf-ml.predicted.csv, where
<reactive cysteine> is the linking cysteine in proximity to which the
warheads were docked. This machine learning based prediction of reactivity
is not currently available for other residues beyond the default cysteine or
other warhead libraries. If you wish to apply the machine learning
algorithm to other residues and/or warhead libraries, please contact
us at support@silcsbio.com. Below are the contents of
each column of <reactive cysteine>.rf-ml.predicted.csv:
In the first column, the warhead names are listed.
In the second and third columns, the LGFEs of the most favorable poses of the corresponding warhead in terms of LGFE (
eq) and ProbRes (rx), respectively, are listed.In the fourth column the ProbRes value of the most favorable pose of the corresponding warhead in terms of ProbRes (
rx) is listed.In the fifth column, the predicted kGSH of the corresponding warhead for the linking cysteine is listed.
In the sixth column, the predicted activity class of the corresponding warhead is listed, where
Lindicates low activity andHindicates high activity.