CHARMM General Force Field (CGenFF)¶
Background¶
The CHARMM General Force Field (CGenFF) covers a wide range of chemical groups present in biomolecules and drug-like molecules, including a large number of heterocyclic scaffolds [1]. The parametrization philosophy behind the force field focuses on quality at the expense of transferability, with the implementation concentrating on an extensible force field.
CGenFF uses the CHARMM additive potential energy function to calculate the energy as a function of the Cartesian coordinates of the system, as shown below.
The bonded or intramolecular part of the potential energy function consists of terms for the bonds, valence angles, torsion or dihedral angles, improper dihedral angles, and a Urey-Bradley term, where \(b_0\), \(\theta_0\), \(\phi_0\), and \(r_{1,3;0}\), respectively, are the bond, angle, improper and Urey-Bradley equilibrium values, the \(K\)’s are the corresponding force constants, and \(n\) and \(\delta\) are the dihedral multiplicity and phase. The nonbonded or intermolecular portion consists of an electrostatic term, with \(q_i\) and \(q_j\) being the respective partial atomic charges on atoms \(i\) and \(j\), and a van der Waals (vdW) term, which is treated by the Lennard-Jones (LJ) 6-12 potential in which \(\epsilon_{ij}\) is the well depth, \(R_{\mathrm{min},ij}\) is the radius, and \(r_{ij}\) is the distance between \(i\) and \(j\).
It is apparent that a simulation on any system of practical interest requires large numbers of parameters. To make the assignment of these parameters practical, force fields require atom types to be assigned to all the atoms in the system, with the parameters associated with combinations of atom types. For instance, the parameter list will contain \(K_\phi\), \(n\), and \(\delta\) values for the dihedral parameters associated with all combinations of four atom types that occur in the molecules supported by the force field. Thus, the first step of assigning parameters for a chemical system is assigning atom types to that system.
CGenFF comes with an algorithm that can automatically assign atom types to a given molecule. The atom typer is rule-based and programmable, making it easy to implement complex atom typing rules and to update the atom typing scheme as the force field grows. Assignment of bonded parameters is based on substituting atom types in the definition of the desired parameter. A penalty is associated with every substitution and the existing parameter with the lowest total penalty is chosen as an approximation for the desired parameter. Charges are assigned using an extended bond-charge increment scheme that is able to capture inductive and mesomeric effects across up to 3 bonds. More details can be found in [2].
Usage¶
CGenFF program accepts Mol2 format files. For an accurate typing, it is
important that all hydrogens are present, in the correct protonation and
tautomeric states, and that the bond orders are correct. Unfortunately, there
are few open source molecular editors that allow the user to visualize and edit
bond orders and save the result as a valid .mol2 file following the Mol2
format standard.
Once Mol2 file is ready, the CGenFF program can be executed by the following command.
${SILCSBIODIR}/programs/cgenff <mol2file>
This produces a stream file in standard output. The output can be redirected to
a .str file. The stream file consists of topology file and the extra
parameter file combined in a single file. It is up to the user to whether to
use the stream file directly or to split the file into two separate files
depending on the needs.
As an example, the adrenalin molecule is parametrized in CGenFF. The example
input and output files can be found in
${SILCSBIO}/examples/cgenff/adrenalin.mol2 and
${SILCSBIO}/examples/cgenff/adrenalin.str, respectively.
The output stream file has two major sections.
* Toppar stream file generated by
* CHARMM General Force Field (CGenFF) program version 1.0.0
* For use with CGenFF version 3.0.1
*
read rtf card append
... topology section ...
END
read param card flex append
... parameter section ...
END
RETURN
In CHARMM, asterisk and exclamation mark represents a comment. If you want to
separate topology and parameter section, you can take out the corresponding
section after read rtf or read param statement to END statement.
Let’s take a look at topology section.
RESI ADRN 0.000 ! param penalty= 113.000 ; charge penalty= 16.607
GROUP ! CHARGE CH_PENALTY
ATOM O1 OG311 -0.649 ! 11.757
ATOM O2 OG311 -0.530 ! 0.000
ATOM O3 OG311 -0.530 ! 0.000
ATOM N NG311 -0.524 ! 16.607
ATOM C1 CG311 0.127 ! 16.171
Above is the beginning of topology section. This section defines the atom names and types, as well as the atomic charge. Each entry is followed by a charge penalty score. The initial atomic charges are assigned based on chemical analogy to the existing model compound. The penalty score becomes high when a good analogy is not found.
Below is an excerpt from the parameter section. When an exact parameter is not found in an existing parameter database, an entry is added based on an analogous parameter. Similar to atomic charges, each entry in parameter is also followed by a penalty score.
ANGLES
CG2R61 CG311 OG311 75.70 110.10 ! ADRN , from CG2R61 CG321 OG311, penalty= 4
CG311 CG321 NG311 43.70 112.20 ! ADRN , from CG331 CG321 NG311, penalty= 1.5
CG321 NG311 CG331 40.50 112.20 5.00 2.42170 ! ADRN , from CG3AM1 NG311 CG3AM1, penalty= 35
High penalty scores indicate that a parameter may be targeted for further optimization. Typically, penalty scores greater than 200 warrant further optimization.
Gromacs readable parameters¶
To get parameters in GROMACS format, execute the following command.
${SILCSBIODIR}/cgenff/cgenff_to_gmx mol=<mol2file>
This command produces three files <mol2file>_gmx.top, <mol2file>_gmx.pdb & <mol2file>_charmm.str.
<mol2file>_gmx.top/pdb contain the GROMACS readable parameters and the coordinate information.
<mol2file>_charmm.str contain CHARMM readable parameters.
CGenFF Parameter Optimizer¶
The CGenFF program performs atom typing and assignment of parameters and charges by analogy in a fully automated fashion. Atom typing is done by a deterministic programmable decision tree. Assignment of bonded parameters is based on substituting atom types in the definition of the desired parameter. A penalty is associated with every substitution and the existing parameter in the current version of the force-field with lowest total penalty is chosen as an approximation for the desired parameter. The penalty score is returned to the user as an indication of approximation needed to obtain the desired parameter. Thus, bonded parameters with higher penalties (typically >200) therefore might require further optimization.
CGenFF optimizer allows for automatic optimization of bonded parameters and thereby improve accuracy by fitting against QM data. Currently, the optimizer works for only rotatable dihedrals. Dihedral scans in QM are performed using Psi4. CGenFF parameters are then fit against this QM data using, LSFitPar, a least-square fitting procedure. If multiple dihedrals are to be optimized, then each dihedral is fitted separately using independent QM and MM scans for each of these dihedrals.
CGenFF Optimizer accepts molecules in mol2 format. All hydrogens must be explicitly defined, all atoms in correct protonation states, and bond-order between atoms correctly defined.
To run the program, set the following environment variables,
export SILCSBIODIR=<silcsbio>
export PSI4DIR=<psi4>
Psi4 package can be obtained from Psi4 download page. The easiest form of installation involves downloading the Binary installer, and running the command:
./Psi4conda-latest-py36-Linux-x86_64.sh
and following the step-by-step guide to installation. After Psi4 has been
installed, set the PSI4DIR variable correctly and proceed with using the
optimizer.
Usage¶
${SILCSBIODIR}/cgenff-optimizer/optimize_cgenff_par mol=<mol2file>
Along with the required argument of mol=<mol2file>, the following additional
parameters can also be set:
- Penalty score :
penalty=<score; default 200>By default, the program identifies rotatable dihedrals with penalties greater than 200 for optimization. This can be changed by adjusting the
penaltyargument, to increase/decrease the list of dihedrals that will be optimized.
- Dihedral step size :
dihedral_step_size=<step size; default 15>Each dihedral identified for optimization is rotated through 360 degrees with a step-size of 15 degrees to generate QM and MM scans. This step-size can be changed by adjusting the
dihedral_step_sizeargument to increase or decrease the resolution of the scan.
- Number of cores used for QM optimization:
nproc=<number of core; default all available cores on workstation/8 cores on HPC>At each scan point for each dihedral, a constrained QM optimization is performed. Psi4 can make use of multiple CPU cores to run this optimization faster. When the optimizer is installed on an HPC, separate jobs are submitted for each of the identified dihedrals simultaneously. Each job requests
nproccores. When the optimizer is installed on a workstation, then each scan point for each dihedral is run one after the other using all the available cores on the workstation by default. Instead of all the available cores, the number of cores used by Psi4 can be changed usingnproc
- Molecular-orbital theory:
mo_theory=<MP2/HF; default HF>Constrained QM optimization for each dihedral at each scan point is performed using Møller–Plesset perturbation theory (MP2). If the user wants to instead use Hartree-Fock (HF), then it can be set using
mo_theory.
Usecase:¶
To illustrate usage of the CGenFF optimizer, following steps delineate the
fitting procedure for ethene_co_isoxazole.mol2 available in the
${SILCSBIODIR}/examples/cgenff/.
${SILCSBIODIR}/cgenff-optimizer/optimize_cgenff_par mol=ethene_co_isoxaole.mol2
The program generates a CHARMM compatible CGenFF stream file named :
ethene_co_isoxazole_optimized.str. Since this molecule does not contain any
dihedral with penalty greater than 200, running the above command will
result in :
Nothing to optimize based on the penalty score threshold of 200
The user can look through the stream file to identify dihedrals that could benefit from optimization:
DIHEDRALS
CG2DC3 CG2DC1 CG2O5 CG2R51 1.4000 2 180.00 ! NONAME.* , from CG2DC3 CG2DC1 CG2O5 OG2D3, penalty= 56.5
HGA4 CG2DC1 CG2O5 CG2R51 0.0000 2 180.00 ! NONAME.* , from HGA4 CG2DC1 CG2O5 OG2D3, penalty= 56.5
CG2DC1 CG2O5 CG2R51 CG2R51 1.5850 2 180.00 ! NONAME.* , from CG2O3 CG2O5 CG2R61 CG2R61, penalty= 126.5
CG2DC1 CG2O5 CG2R51 OG2R50 1.5850 2 180.00 ! NONAME.* , from CG2O3 CG2O5 CG2R61 CG2R61, penalty= 176
OG2D3 CG2O5 CG2R51 CG2R51 1.5850 2 180.00 ! NONAME.* , from OG2D3 CG2O5 CG2R61 CG2R61, penalty= 93.5
OG2D3 CG2O5 CG2R51 OG2R50 1.5850 2 180.00 ! NONAME.* , from OG2D3 CG2O5 CG2R61 CG2R61, penalty= 143
CG2O5 CG2R51 CG2R51 CG2R52 0.0000 2 180.00 ! NONAME.* , from CG2O1 CG2R51 CG2R51 CG2R52, penalty= 3
CG2O5 CG2R51 CG2R51 HGR51 1.0000 2 180.00 ! NONAME.* , from CG2R51 CG2R51 CG2R51 HGR51, penalty= 66.5
CG2O5 CG2R51 OG2R50 NG2R50 8.5000 2 180.00 ! NONAME.* , from CG2R51 CG2R51 OG2R50 NG2R50, penalty= 66.5
If the user now wants to fit CG2DC1 CG2O5 CG2R51 OG2R50, which has received a penalty score of 176,
then the program can be re-run by lowering the penalty threshold to 175:
${SILCSBIODIR}/cgenff-optimizer/optimize_cgenff_par mol=ethene_co_isoxazole.mol2 penalty=175
This will result in:
#############################################
for dihedral = CG2DC1 CG2O5 CG2R51 OG2R50, parameter penalty = 176.000000;
#############################################
Will be optimizating the parameters of the above listed dihedral(s)
It is possible that doing so will lead to more than just the dihedral of
interest to be fitted. If however, the user wants to specifically fit only
CG2DC1 CG2O5 CG2R51 OG2R50, then instead of lowering the penalty threshold,
the user can manually supply this dihedral on prompt:
${SILCSBIODIR}/cgenff=optimizer/optimize_cgenff_par mol=ethene_co_isoxazole.mol2
will result in :
CHARMM General Force Field (CGenFF) program version 2.1.0
released the 27th of October 2016
Copyright (C) 2017 SilcBio LLC
and University of Maryland, School of Pharmacy. All Rights Reserved.
Now processing molecule sulf ...
#############################################
#############################################
Nothing to optimize based on the penalty score threshold of 200
Do you want to perform optimization with anymore dihedrals? [Y/N; default N]
To add to the list, type Y, and then supply dihedral(s) in the format of
(AtomType1 AtomType2 AtomType3 AtomType4). The program then summarizes the
updated-list:
List updated; will be attempting optimization with the following dihedrals
#############################################
CG2DC1 CG2O5 CG2R51 OG2R50
#############################################
In general, to identify the atom-typing corresponding to the atoms in a
dihedral, the user can look at the last column of
<mol>_cgenff_atomtypes.pdb. In this case,
ethene_co_isoxazole_cgenff_atomtypes.pdb
ATOM 1 C NONA 1 0.759 0.680 1.188 1.00 0.00 CG2R52
ATOM 2 H NONA 1 1.645 1.222 1.497 1.00 0.00 HGR52
ATOM 3 C NONA 1 -0.353 1.220 0.530 1.00 0.00 CG2R51
ATOM 4 H NONA 1 -0.508 2.244 0.227 1.00 0.00 HGR51
ATOM 5 C NONA 1 -1.205 0.177 0.349 1.00 0.00 CG2R51
ATOM 6 C NONA 1 -2.542 0.163 -0.296 1.00 0.00 CG2O5
ATOM 7 C NONA 1 -3.305 -1.129 -0.378 1.00 0.00 CG2DC1
ATOM 8 H NONA 1 -2.855 -2.041 0.043 1.00 0.00 HGA4
ATOM 9 C2 NONA 1 -4.530 -1.255 -0.944 1.00 0.00 CG2DC3
ATOM 10 H21 NONA 1 -5.050 -2.226 -0.980 1.00 0.00 HGA5
ATOM 11 H22 NONA 1 -5.045 -0.383 -1.387 1.00 0.00 HGA5
ATOM 12 O1 NONA 1 -3.016 1.204 -0.756 1.00 0.00 OG2D3
ATOM 13 O NONA 1 -0.636 -0.949 0.875 1.00 0.00 OG2R50
ATOM 14 N NONA 1 0.613 -0.617 1.406 1.00 0.00 NG2R50
Following the identification of the dihedrals to the fitted, the program first performs a complete minimization of molecule using the initially obtained CGenFF parameters. Next, each of the selected dihedrals are independently rotated by 360 degrees through the specified step-size. Separate Psi4 jobs for constrained QM optimization around the specified dihedral are then performed either serially, one-by-one when running on a workstation, or parallely when running on a HPC cluster.
The program waits on the QM jobs to finish. After the completion of the QM jobs,
MM energies are calculated for each of the scan-points. Both the QM and MM
energies are then used by the lsfitpar program to fit the dihedral parameter.
Updated parameters are saved here : <mol>_optimized.str.
Additionally, the dihedral scans from QM, MM can be visualized using the image
files : qm_mm_*.png. Note, if the installed location does not contain
gnuplot, or gnuplot linked against png library, then the user can copy the 4plot
directory to a location that contains gnuplot and run the gnuplot files there.
For instance, for the ethene_co_isoxazole, the following is an example of
dihedral scans obtained from using the optimization procedure:
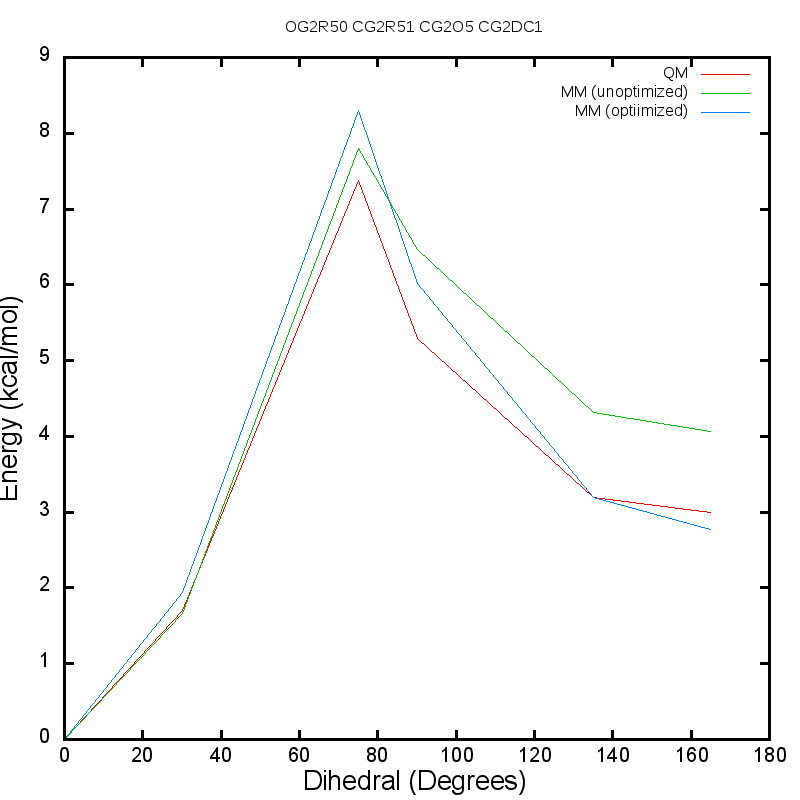
Initial guess for dihedral CG2DC1 CG2O5 CG2R51 OG2R50 :
CG2DC1 CG2O5 CG2R51 OG2R50 1.5850 2 180.00 ! RMSE = 0.468999
After fitting,
CG2DC1 CG2O5 CG2R51 OG2R50 0.5728 2 0.00 ! RMSE = 0.111323