Ligand docking and ranking on the GFE FragMaps¶
An SILCS-MC simulation involves Monte-Carlo (MC) sampling of the ligand in translational, rotational and torsional space in the field of FragMaps. The energy of a ligand conformation is evaluated by the combination of CGenFF intramolecular energies and LGFE (Ligand Grid Free Energy) score, which is the sum of atomic GFEs. The ligand doesn’t “see” the protein, but the exclusion map prevents it from sampling the region where no probe or water molecules visited during the SILCS simulations, i.e., interior of the protein. This allows for rapid docking of the ligand while accounting for protein flexibility in a mean- field like fashion as that information is embedded in the FragMaps and the exclusion map. For more details see [5].
Two SILCS-MC presets are made available to the user: a) exhaustive pocket sampling, and b) local sampling. In the following sections, we will describe the parameters and procedures to perform SILCS-MC runs. Each of the preset parameters can be modified to make a new customized SILCS-MC protocol. The instruction on customizing these parameters/procedure will be given at the end of the document.
SILCS-MC Presets¶
Exhaustive pocket sampling¶
As the name suggests, this approach enables an exhaustive sampling of a ligand conformation in a given pocket to determine a most favorable binding pose based on LGFE scoring. The pocket here is predefined as a 10 Å sphere, and the center of the pocket is either defined as the center of the supplied ligand molecule or explicitly given by the user. This protocol involves running five independent MC runs with the ligand.
This protocol is recommended to sample and score ligands with diverse chemotype and unknown binding poses. When the pose of a parent ligand is known and MC- SILCS evaluations are to be performed over a congeneric series, another procedure called “local search” is recommended (see below).
To setup and run this SILCS-MC procedure, create a directory containing all the
ligands to be scored using the SILCS FragMaps. Each ligand can be stored as a
separate .mol2 or .sdf files. Alternatively, all the ligands can be
combined into a single .sdf file.
${SILCSBIODIR}/silcs-mc/1_run_silcsmc_exhaustive \
prot=<prot pdb> \
ligdir=<directory containing ligand mol2/sdf> \
mapsdir=<directory containing SILCS FragMaps> \
center="x,y,z"
To learn more about the command-line options, run:
${SILCSBIODIR}/silcs-mc/1_run_silcsmc_exhaustive
- This procedure of exhaustive sampling is spawned as five independent single- core serial jobs per ligand and take an average between 30 minutes to one hour to complete for a single ligand (depending on system architecture).
- Each MC run involves a maximum of 250 cycles (and a minimum of 50 cycles) of MC/SA sampling of the ligand within a 10 Å sphere. When no sphere center is provided by the user, the center-of-mass of the ligand is treated as the center of the sphere.
- Each of these 250 cycles comprises 10,000 steps of MC at a high temperature followed by a 40,000 steps of Simulated Annealing (SA) towards a lower temperature.
- At the beginning of each cycle, the ligand will be reoriented within the predefined sphere. When no sphere center is defined by the user, ligand orientation defined in the Mol2/sd file will be used instead as a starting pose at the start of every cycle.
- The MC sampling has three types of moves: i) molecular translations with a
maximum step size of 1 Å, ii) molecular rotation with a maximum step size of
180 degrees and, and iii) intramolecular dihedral rotations with a maximum
step size of 180 degrees. For the intramolecular dihedral rotations, only the
rotatable dihedral angles are selected for MC moves. The lowest LGFE scoring
pose from the MC sampling is used as starting pose in the following SA
sampling. The SA sampling also involves the same three types of moves, but
with a smaller step size compared to the MC sampling: i) molecular
translations with a maximum step size of 0.2 Å, ii) molecular rotation with a
maximum step size of 9 degrees and, iii) intramolecular dihedral rotations
with a maximum step size of 9 degrees. The lowest LGFE scoring pose from the
SA is saved in the multi-frame PDB file (
3_silcsmc/<run>/pdb/<lig>_mc.<run>.pdb). - In each run, after a minimum of 50 MC/SA cycles, if the LGFE score difference between the top three poses (defined by lowest LGFE scores) are less than 0.5 kcal/mol, then that run is considered converged and terminated.
- If the top three scored poses are separated by more 0.5 kcal/mol, the MC/SA procedure continues until the convergence criteria is met. After a maximum of 250 MC/SA cycle, the runs is terminated regardless of convergence criteria.
- After all the five runs are finished, the lowest LGFE (best scored) pose among
all the five runs is retrieved for visualization
(
3_silcsmc/minconfpdb/<lig>_silcsmc.minconf.pdb).
Local Sampling¶
This protocol enables SILCS-MC driven local-sampling of a ligand conformation starting from a user-defined pose in the protein pocket. The pocket is defined as the center-of-mass of the ligand supplied by the user. The orientation found in the same user-supplied ligand file is used for the initial ligand pose.
This protocol is designed to only sample near the conformations supplied by the user, therefore this protocol is recommended when there is a high confidence in the parent ligand and/or SILCS-MC evaluations are to be performed over a congeneric series.
To setup and run this SILCS-MC procedure, create a directory containing all the
ligands to be scored using the SILCS FragMaps. Each ligand can be stored as a
separate .mol2 or .sdf files. Alternatively, all the ligands can be
combined into a single .sdf file.
${SILCSBIODIR}/silcs-mc/1_run_silcsmc_local prot=<prot pdb> \
ligdir=<directory containing ligand mol2/sdf> \
mapsdir=<directory containing SILCS FragMaps>
To learn more about the command-line options, run:
${SILCSBIODIR}/silcs-mc/1_run_silcsmc_local -h
- This procedure of exhaustive sampling is spawned as five independent single- core serial jobs per ligand.
- Each MC run involves a maximum of 10 cycles of MC/SA sampling of the ligand within a 1 Å sphere. The center of the sphere is defined as the center-of-mass of the ligand.
- Each of the 10 cycles comprises 100 steps of MC sampling at a high temperature, followed by 1000 steps of SA sampling towards a low temperature.
- At the beginning of each cycle, the ligand orientation/conformation will be reset by the ones found in the user supplied Mol2/sd file.
- The MC sampling has three types of moves: i) molecular translations with a
maximum step size of 1 Å, ii) molecular rotation with a maximum step size of
180 degrees and, and iii) intramolecular dihedral rotations with a maximum
step size of 180 degrees. For the intramolecular dihedral rotations, only the
rotatable dihedral angles are selected for MC moves. The lowest LGFE scoring
pose from the MC sampling is used as starting pose in the following SA
sampling. The SA sampling also involves the same three types of moves, but
with a smaller step size compared to the MC sampling: i) molecular
translations with a maximum step size of 0.2 Å, ii) molecular rotation with a
maximum step size of 9 degrees and, iii) intramolecular dihedral rotations
with a maximum step size of 9 degrees. The lowest LGFE scoring pose from the
SA is saved in the multi-frame PDB file (
3_silcsmc/<run>/pdb/<lig>_mc.<run>.pdb). - After all the five runs are finished, the lowest LGFE (best scored) pose among
all the five runs is retrieved for visualization
(
3_silcsmc/minconfpdb/<lig>_silcsmc.minconf.pdb).
User-defined Protocols¶
Apart from the two presets, users can define their own protocols to drive
SILCS- MC sampling of ligand(s) in the field of FragMaps. To do so, copy the
${SILCSBIODIR}/templates/silcs-mc/params_custom.tmpl to the location where
you plan to setup the SILCS-MC calculation and edit this file. The parameters
in the bracket, e.g., <parameter>, will be replaced later when the program
is executed.
The parameters in the angle brackets, e.g., <parameter>, will be replaced by
the program to a default value when the program is executed. User will be
responsible for providing and making sure to using:
${SILCSBIODIR}/programs/silcs_mc -h
After the desired parameters are set in the params_custom.tmpl, use the
following command to setup and run SILCS-MC procedure. Each ligand can be
stored as a separate .mol2 or .sdf file. Alternatively, all the ligands
can be combined into a single .sdf file.
${SILCSBIODIR}/silcs-mc/1_run_silcsmc_custom prot=<prot pdb> \
ligdir=<directory containing ligand mol2/sdf> \
mapsdir=<directory containing SILCS FragMaps> \
paramsfile=<params_custom.tmpl>
Additionally, the number of runs that will be spawned can also be modified, by
using another command-line parameter, totruns.
${SILCSBIODIR}/silcs-mc/1_run_silcsmc_custom prot=<prot pdb> \
ligdir=<directory containing ligand mol2/sdf>
mapsdir=<directory containing SILCS FragMaps>
paramsfile=<params_custom.tmpl>
totruns=<# of runs>
See below for the detailed description of parameters that need to be defined in
the params_custom.tmpl file.
CGENFF_RULES <cgenff rules_file>(required)This file is needed by the internal CGenFF library to determine the correct force-field parameters for the ligand. The default value is
${SILCSBIODIR}/data/cgenff.rulesCGENFF_PAR <cgenff parameter file>(required)Along with the CGENFF_RULES file, this file is needed by the internal CGenFF library to determine the correct force-field parameters for the ligand. The default value is
${SILCSBIODIR}/data/par_all36_cgenff.prmSILCS_RULES <silcs rules file>(required)This rule file is used to map the different atoms in the ligand to the corresponding SILCS FragMap types. This mapping is used to determine the appropriate “field” that will be applied to the different atoms in the ligand when attempting an MC-move. The default value is
${SILCSBIODIR}/data/silcs_classification_rules_feb16_generic.datAnother variant of this rule file is available at
${SILCSBIODIR}/data/silcs_classification_rules_feb16_specific.dat.When
silcs_classification_rules_feb16_generic.datis used, ligand atoms are assigned using six generic classifications in mapping them back to the FragMaps:APOLAR,HBDON,HBACC,MEOO,ACEO, andMAMN. All the atoms in the ligand fall into one of these six categories.When
silcs_classification_rules_feb16_specific.datis used, ligand atoms are assigned using 13 more specific classifications in mapping them back to FragMaps:BENC,PRPC,AALO,MEOO,FORN,FORO,IMIN,IMIH,ACEO,MAMN,APOLAR,HBDONandHBACC. For instance, aromatic ring carbons and aliphatic linker carbons are distinguished asBENCandPRPC, respectively here, while they are grouped asAPOLARatoms in the generic rule file.GFE_CAP <default: 3.0>Maximum unfavorable GFE (kcal/mol) accounted in the MC calculation.
RDIE <default: true>When true, the distance dependent dielectric (RDIE) scheme is used to treat intramolecular electrostatics. When false, CDIE (constant dielectric scheme) is used.
DIELEC_CONST <default: 4>Dielectric constant used in the intramolecular electrostatic interactions calculations.
MIN_STEPS <default: 10000>Maximum number of steps of minimization performed using the steepest-descent algorithm with the ligand, before initiating MC simulation.
EMTOL <default: 0.01>Minimization is converged when the diff in total energy (totE) across the last 10 steps is smaller than this value. Once this criteria is satisfied minimization terminates.
MC_MOVE_RANGE <default:1.0 180.0 180.0>.Max range of translation, rigid body rotation and dihedral rotation per step of MC simulation.
MC_PRNT_FRQ <default: 0>Number of intermediate steps of MC to be written into OUTMCPDBFILE.
MC_STEPS <default: 10000>Number of steps of MC simulation to be performed per cycle.
SIM_ANNEAL_MOVE_RANGE <default:0.2 9.0 9.0>Max range of translation, rigid body rotation and dihedral rotation per step of simulated annealing simulation after MC simulation.
SIM_ANNEAL_STEPS <default: 40000>Number of steps of simulated annealing to be performed per cycle.
INIT_RUNS <default: 50>Number of MC/SA cycles before initiating checks for convergence.
NUM_TOL <default: 3>Number of top-scoring cycles with differences in LGFE less than DELTAE_TOLERANCE, before this simulation (run) is considered converged.
DELTAE_TOLERANCE <default: 0.5>When differences in LGFE of NUM_TOL most-favorable cycles are less than this defined tolerance value, convergence is reached and the program exits
DELTAE_BUFF <default: 10>Progression of MC+SA from one cycle to next is such that LGFE (of lowest conf) from MC should be less than (prev_min+deltae_buff). This ensures that N cycles are proceeding towards a minimum lower than that previously discovered lowest energy conformation.
TOTE_CRITERIA <default: false>When true, instead of LGFE, total energy (totE) of the system is used for convergence checks. Useful when running vacuum-phase MC simulations of the ligand.
TOT_RUNS <default: 250>Maximum number of MC simulation cycles. The program terminates if the
DELTAE_TOLERANCEcriteria is satisfied before reachingTOT_RUNS. Alternately, even if theDELTAE_TOLERANCEcriteria is not satisfied when the number of cycles executed reachesTOT_RUNS, the program terminates.RANDOM_SEED: <default: system-time>Seed used in MC simulation. When not set, system-time is used as a seed.
SIMULATION_CENTER: <x,y,z>Cartesian coordinates of where the MC simulation should be performed.
SIMULATION_RADIUS: <default: 1.0 A>Radius of the sphere within which MC simulation will be performed.
RANDOM_INIT_ORIENT: <true/false>When set to
TRUE,SIMULATION_CENTERshould also be set. The ligand is placed within a sphere of size,SIMULATION_RADIUS, in a random orientation and a conformationWhen set to
FALSE, then the center-of-mass of the ligand is used as the center for the MC simulation. This is useful when the ligand pose in the pocket is well-known.ATOM_TO_RESTRAIN: <atom number in sd/mol2>When set, a spherical potential is applied to restrained the defined atom within the sphere during MC moves. This enables geometrically restraining a particular pharmacophore feature. Note, when using this feature, supply the full molecule with explicit hydrogens already added. Also, random pocket pose and placement using
RANDOM_INIT_ORIENTtrue is incompatible.When not set, the entire molecule is free to rotate/move/translate
ATOM_RESTRAINT_CENTER: <x,y,z>To be used in conjunction with
ATOM_TO_RESTRAINoption. This value is used to defined the center of the spherical potential.ATOM_RESTRAINT_RADIUS: <default: 1.0 A>To be used in conjunction with
ATOM_TO_RESTRAINoption. This value is used to defined the radius of the spherical potential. When not defined, then a default of 1 A is used.OUTRMSDFILE <output RMSD file>This file stores the RMSD and LGFE of the lowest energy conformation from each run of the MC/SA simulation. To be used in conjunction with
RANDOM_INIT_ORIENTset totrue.CLUSTER_RADIUS <default: 0.6 A>Used clustering for ligand binding poses.
OUTCLUSTPDBFILE: <output PDB file>This file stores representative cluster conformations.
LIGAND_SDF: <ligand sdf>When ligands are in an
.sdfile, use this option. Only if no sdf is supplied from command-line, this line will be read.LIGAND_MOL2: <ligand mol2>When ligands are in mol2 file, use this option. Only if no mol2 is supplied from command-line, this line will be read
One of these parameters (LIGAND_MOL or LIGAND_SDF) need to be set correctly for SILCS-MC simulation to proceed. Interchanging
LIGAND_MOL2andLIGAND_SDFfor the wrong file-types lead to SILCS-MC simulation to not proceed.SILCSMAP <MapType> <map name>(required)Multiple
SILCSMAPflags can be defined, with each flag pointing to one file. Standard SILCS FragMaps of the following<MapType>should be included with the below keywords:- EXCL - exclusion map
- NCLA - zero map for non-classified ligand atoms
- BENC - benzene carbon
- PRPC - propane carbon
- MEOO - methanol alcohol oxygen
- FORN - formamide nitrogen
- FORO - formamide oxygen
- MAMN - methyl ammonium nitrogen
- ACEO - acetate oxygens
- AALO - acetaldehyde oxygen
- IMINH - imidazole donor nitrogen
- IMIN - imidazole acceptor nitrogen
- APOLAR - generic non polar : green
- HBDON - generic donor: blue
- HBACC - generic acceptor: red
OUTMCPDBFILE <output PDB file>(required)This file stores the lowest energy conformation from each cycle of the MC/SA simulation.
OUTMCLOGFILE <output log file>This file stores the energy statistics of the lowest energy conformation from each cycle of the MC/SA simulation.
OUTMCPDBFILEPREFIX <output PDB file(s)>When reading SD files (with one or more molecules), this file(s) store the lowest energy conformation from each cycle of the MC/SA simulation. Filenames are appended with the name of the ligand(s)
OUTMCLOGFILEPREFIX <output log file(s)>When reading SD files (with one or more molecules), this file(s) stores the energy statistics of the lowest energy conformation from each run of the MC/SA simulation. Filenames are appended with the name of the ligand(s)
If
LIGAND_SDFis defined, thenOUTMCPDBFILEPREFIXandOUTMCLOGFILEPREFIXneed to be defined and need to point to the right files for SILCS-MC to proceed.Note
Ligand related parameters such as
LIGAND_MOL2/LIGAND_SDF,OUTMCPDBFILE/OUTMCLOGFILE, need not be manually set. The script1_run_silcsmc_customwill set these parameters appropriately for each of the ligand defined in theligdirdirectory.
LGFE and Best Pose retrieval¶
Once the SILCS-MC simulation is finished, retrieve the LGFE scores for each of the ligands that have been subjected to SILCS-MC using:
${SILCSBIODIR}/silcs-mc/2_calc_lgfe_min_avg_sd ligdir=<directory containing lig mol2>
An example of the output of this script is:
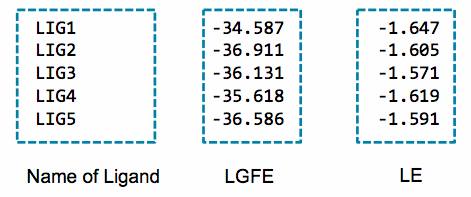
An alternative to the LGFE score is the ligand efficiency (LE). The LE is calculated as the LGFE score divided by the number of heavy atoms in each ligand.
Binding pocket identification¶
Please inquire for more information on this capability.
Excipient design for biologics¶
Please inquire for more information on this capability.