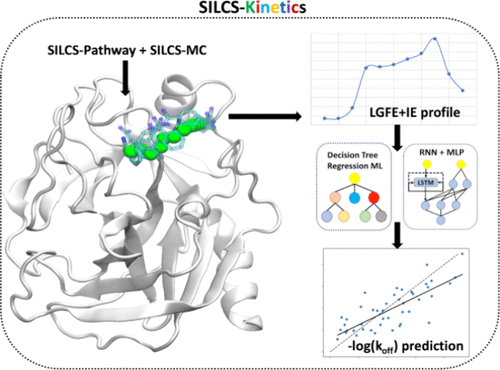
SILCS-Kinetics
Background
SILCS-Kinetics is a computational workflow designed to efficiently predict ligand dissociation kinetics, specifically the off rate (koff), which is often more relevant to drug efficacy than binding affinity alone. Traditional approaches rely on enhanced-sampling molecular dynamics (MD) simulations, which are computationally intensive and less practical for evaluating large ligand libraries in drug design.
To address this, SILCS-Kinetics combines physics-based and machine learning (ML) methodologies. The workflow uses the Site Identification by Ligand Competitive Saturation (SILCS) method to enumerate potential ligand dissociation pathways and calculate free-energy profiles along those pathways. These profiles, together with molecular properties, serve as features for training ML models—including tree-based and neural network approaches—to predict koff values.
The protocol has been developed and validated on a diverse set of 329 ligands across 13 proteins, demonstrating the robustness and efficiency of the ML workflow built upon SILCS-derived free-energy profiles. SILCS-Kinetics provides a powerful and scalable tool for drug design, enabling rapid quantitative estimates of ligand dissociation kinetics and atomic or functional group contributions to unbinding events. Additional details on the development and validation of SILCS-Kinetics can be found in the original publication [25].
SILCS-Kinetics Using the SilcsBio CLI
Cluster Generation and SILCS-MC Slab Preparation
This step prepares and runs the cluster-generation phase of the SILCS-Kinetics workflow for a given protein structure. It identifies ligand trajectory clusters using a specified cutoff, then creates and organizes SILCS-MC input files for every cluster–ligand pair.
Usage
${SILCSBIODIR}/silcs-kinetics/1_gen_silcsmc_slab prot=<protein PDB file>Required parameters
prot: Path to the protein PDB file.
Optional parameters
outputdir: Output directory for results (default: 5_kinetics).pathwaydir: Path to the silcs-pathway output directory (default: 4_pathway).paramsfile: Path to SILCS-MC parameters template file.cutoff: Minimum number of members in a cluster to be included in kinetics runs (minimum: 2) (default: 2).
Output
SILCS-MC input files for each cluster–ligand pair in pathclusters/.
Log files and cluster information.
Notes
The script checks for valid input files and parameters.
Clusters with fewer members than the cutoff are excluded.
SILCS-MC Job Submission
This step submits SILCS-MC simulations for all cluster–ligand pairs identified in the previous step.
Usage
- ::
${SILCSBIODIR}/silcs-kinetics/2_submit_jobs prot=<protein PDB file> ligdir=<ligand directory> ${SILCSBIODIR}/silcs-kinetics/2_submit_jobs prot=<protein PDB file> ligdir=<ligand directory>
Required parameters
prot: Path to the protein PDB file.ligdir: Directory containing ligand mol2/sdf files.
Optional parameters
outputdir: Output directory for results (default: 5_kinetics).sdfile: SD file to overwrite ligdir.
Output
SILCS-MC simulation outputs for each cluster–ligand pair.
Log files and job status information.
Notes
The script checks for valid input files and parameters.
Output directory must be prepared by step 1.
Energy Extraction and Barrier Calculation
This step processes the output from SILCS-MC simulations to extract energy terms and calculate barriers for each ligand–protein cluster. It also prepares data files for machine learning predictions.
Usage
- ::
${SILCSBIODIR}/silcs-kinetics/3_extract_energy prot=<protein PDB file> ${SILCSBIODIR}/silcs-kinetics/3_extract_energy prot=<protein PDB file>
Required parameters
prot: Path to the protein PDB file.
Optional parameters
outputdir: Output directory for results (default: 5_kinetics).
Output
Extracted energy terms and barrier calculations for each cluster–ligand pair.
Data files for ML predictions.
Notes
The script checks for valid input files and parameters.
Output directory must contain successful SILCS-MC results.
Adds script extract_pathway_lgfe.py to outputdir which given a pc# will extract the ligand grid free energy (LGFE) along the pathway for that cluster.
Usage
- ::
${SILCSBIODIR}/silcs-kinetics/4_ml_inference prot=<protein PDB file> python extract_pathway_lgfe.py –pc N [–ligand ligand1 ligand2 …] [–save_plot true/false] [–show_plot true/false]
Example
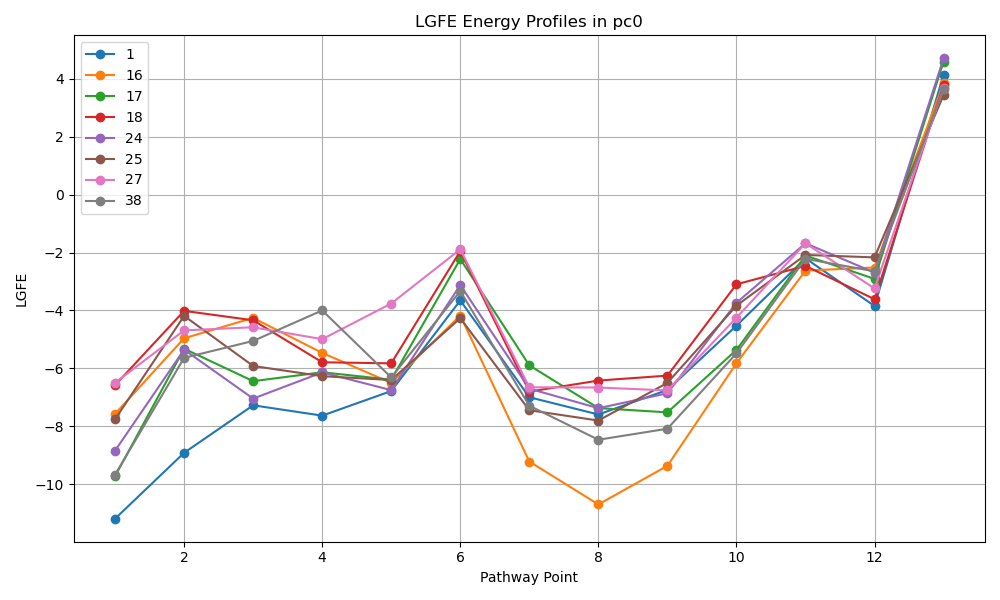
Machine Learning Inference
This step performs the final ML inference, extracting features from SILCS-MC outputs and applying trained models to predict –log(koff) values for each ligand–protein cluster pair.
Usage
- ::
${SILCSBIODIR}/silcs-kinetics/4_ml_inference prot=<protein PDB file>
Required parameters
prot: Path to the protein PDB file.
Optional parameters
outputdir: Output directory for results (default: 5_kinetics).ml_model: ML model type to use for inference (RF, RNN, or both; default: RF).python: Path to Python executable.
Output
Predicted –log(koff) values for each ligand–protein cluster pair.
ML inference log files and results.
Notes
The script checks for valid input files, parameters, and model type.
Output directory must contain successful energy extraction results.
Training the ML Models using the CLI
Scripts are provided to train the ML models used in the SILCS-Kinetics workflow. These scripts can be run from the command line and require specific input data for training. These scripts are located in the utils directory:
${SILCSBIODIR}/utils/python/silcs-kinetics/ml_inference/{tree/rnnmlp}/train/training/
The structured input data for each model varies slightly but both include the silcs-kinetics energy terms, physical properties and experimental data. For the RF model, the input data is a CSV file with the following columns:
ligand_id
Sum of the LGFE barriers
Sum of the IE differences
LGFESMS
MW
Number of rotatable bonds (NRot)
Experimental –log(koff)
Numbers 1-4 are dervived from SILCS-Kinetics and can be found in the ml.bar file generated in step 3 of the workflow. Number 5-6 are physical properties which can be calculated independently or using an included python script.
python $SILCSBIODIR/utils/python/silcs-kinetics/sdf2smi.py
python $SILCSBIODIR/utils/python/silcs-kinetics/physchem_calc.py
Alternatively, you can run your data through step 4 of the workflow which will run the above scripts and generates all.prop file which contains the required properties for all excipients or in the 4_ml/tree directory is a file ml-rf.csv which contains the properly structured input for all ligands.
Training for the RNN-MLP model requires a different input format. The train data is split into two files and a directory. In order to generate the required input to generate this input you should run the RNN-MLP model described above. After running the RNN-MLP model, the required input file will appear in the 5_kinetics folder. In the run directory (where 5_kinetics/ directory is located) you should add a file neg-logkoff.exp which should have two columns excipient name and experimental -log(koff) value separated by a tab. A script called prep_rnn_input.sh can be used to prepare the input for a specific protein for the RNN-MLP model. This script is located at:
${SILCSBIODIR}/utils/python/silcs-kinetics/ml_inference/rnnmlp/train/training/prep_rnn_input.sh
After preparing the files ml-rnn-input.csv for each protein in the test/train set all these files should be concatented into path.train, path.test, and path.val for your training, testing and validation set. A script called rnnmlp_preprocessing.sh is provided to take the path files generated and normalize them to prepare for training. This script is located at:
${SILCSBIODIR}/utils/python/silcs-kinetics/ml_inference/rnnmlp/train/training/rnnmlp_preprocessing.sh
Note this script will need run for separately for the test, train and validation sets. After running this script you will have the properly formatted input for training the RNN-MLP model. These input files should then be moved to the train/data directory located at:
${SILCSBIODIR}/utils/python/silcs-kinetics/ml_inference/rnnmlp/train/data/
The scripts for training the RNN-MLP model are located in the following directory:
${SILCSBIODIR}/utils/python/silcs-kinetics/ml_inference/rnnmlp/train/training/rnnmlp_train.py
${SILCSBIODIR}/utils/python/silcs-kinetics/ml_inference/rnnmlp/train/training/rnnmlp_tuner.py
The tuner script will retune hyperparameters and train a new model, while the train script will train a new model using teh default hyperparameters from original model tuning.
Instructions for training the models can be found in the README files in the tree/rnnmlp directories within the $SILCSBIODIR/utils/python/silcs-kinetics/ml_inference/ directory.