CGenFF Program¶
Background¶
The CHARMM General Force Field (CGenFF) covers a wide range of chemical groups present in biomolecules and drug-like molecules, including a large number of heterocyclic scaffolds [3]. The parametrization philosophy behind the force field focuses on quality at the expense of transferability, with the implementation concentrating on an extensible force field.
The CGenFF program uses the CHARMM additive potential energy function to calculate the energy as a function of the Cartesian coordinates of the system, as shown below.
The bonded portion of the potential energy function consists of terms for the bonds, valence angles, torsion or dihedral angles, improper dihedral angles, and a Urey-Bradley term, where \(b_0\), \(\theta_0\), \(\phi_0\), and \(r_{1,3;0}\), respectively, are the bond, angle, improper and Urey-Bradley equilibrium values, the \(K\)’s are the corresponding force constants, and \(n\) and \(\delta\) are the dihedral multiplicity and phase. The nonbonded portion consists of an electrostatic term, with \(q_i\) and \(q_j\) being the respective partial atomic charges on atoms \(i\) and \(j\), and a van der Waals (vdW) term, which is treated by the Lennard-Jones (LJ) 6-12 potential in which \(\epsilon_{ij}\) is the well depth, \(R_{\mathrm{min},ij}\) is the radius, and \(r_{ij}\) is the distance between atoms \(i\) and \(j\).
Simulation of typical molecular systems of interest requires large numbers of parameters. To make the assignment of these parameters practical, force fields require atom types to be assigned to all the atoms in the system, with the parameters associated with combinations of atom types. For instance, the parameter list will contain \(K_\phi\), \(n\), and \(\delta\) values for the dihedral parameters associated with all sequentially bonded combinations of four atom types that occur in the molecules supported by the force field. Thus, the first step of assigning parameters for a chemical system is assigning atom types to that system.
The CGenFF program employs an algorithm that can automatically assign atom types to a given molecule. The atom typer is rule-based and programmable, making it easy to implement complex atom typing rules and to update the atom typing scheme as the force field grows. In the event that the CGenFF force field file does not explicitly contain a bonded parameter required for simulating the molecule, the CGenFF program will assign bonded parameters by substituting analogous atom types in the definition of the desired parameter. A penalty is associated with every substitution and the existing parameter with the lowest total penalty is chosen as an approximation for the desired parameter. Charges are assigned using an extended bond-charge increment scheme that is able to capture inductive and mesomeric effects across up to 3 bonds. More details can be found in [4].
The CGenFF Suite provides the CGenFF program in both the SilcBio GUI and CLI formats. The SilcsBio GUI provides an intuitive option for users to parametrize their ligands. For advanced users, the CLI allows for more customization and additional features. The usage of CGenFF with the SilcsBio GUI and CLI are described below.
CGenFF Using the SilcsBio GUI¶
Enter the CGenFF Suite:
As of Release v 2024.1, the SilcsBio GUI allows users to use CGenFF. The CGenFF Suite is acessed by selecting from the menu bar.
Begin a new CGenFF project:
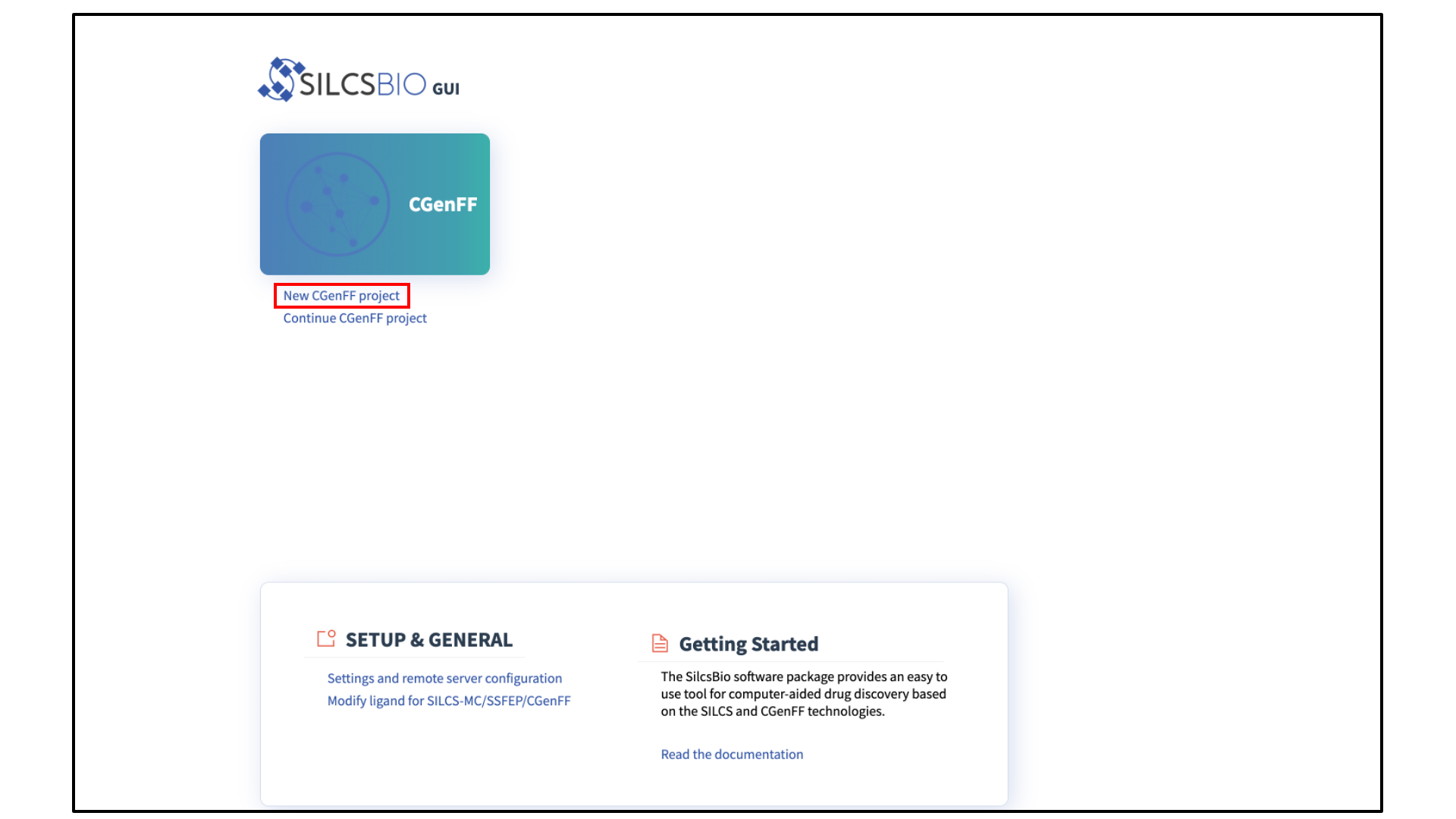
Select New CGenFF project from the Home page.
Enter a project name select the remote server:
Enter a project name and select the remote server where the CGenFF job will run. Input and output files from the CGenFF job will be stored on this server. For information on setting up the remote server, please refer to Remote Server Setup. Next, select a molecule Mol2 file. As described in File and Directory Selection, choose a file from your local computer (“localhost”) or from any server you had configured through the Remote Server Setup process. For correct atom typing, it is important that all hydrogens are present, the system has correct protonation and tautomeric states, and that the bond orders are correct.
Set up the CGenFF job:
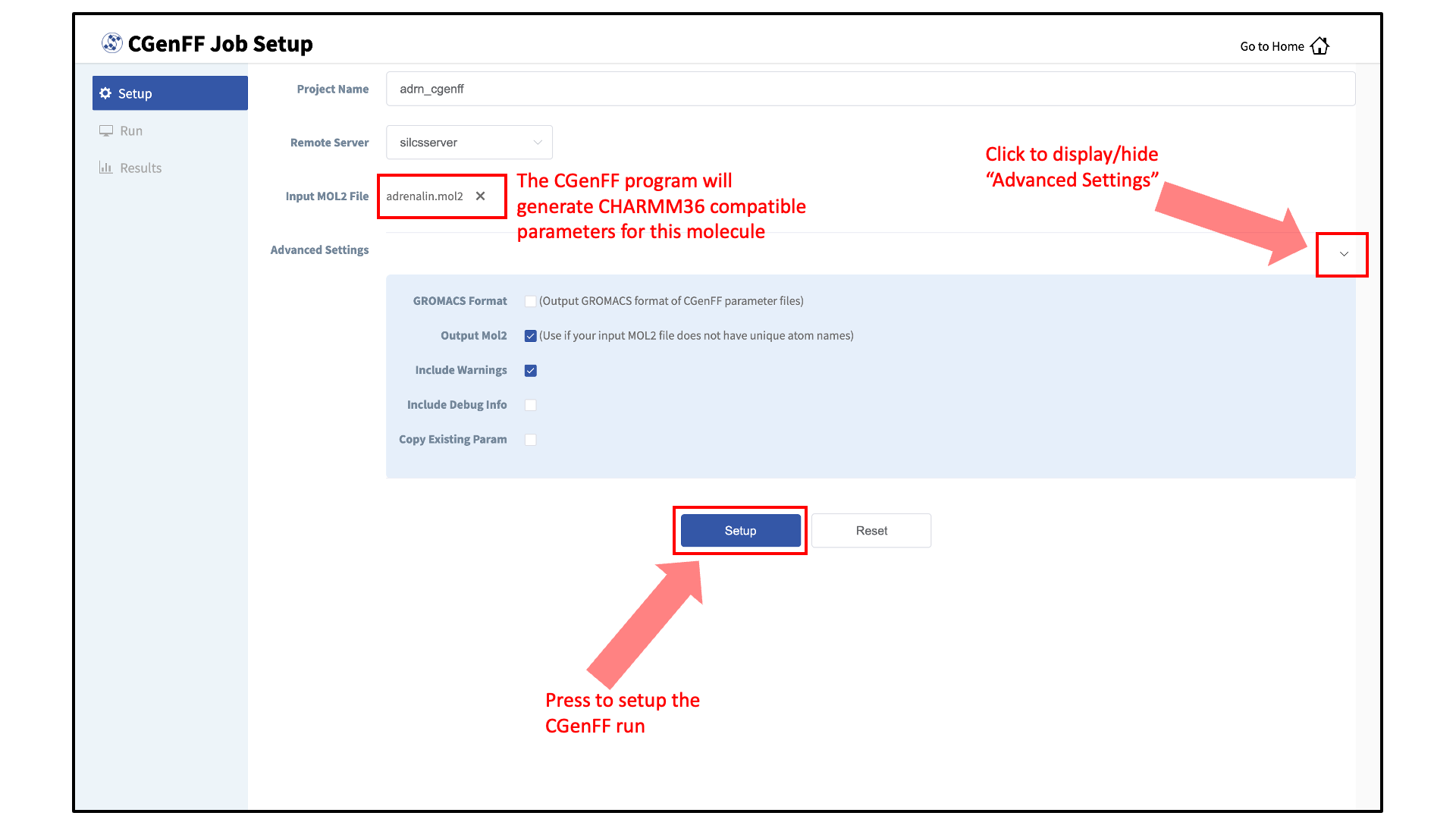
Under “Advanced Settings”, desired information can be specified in the CGenFF output through selecting the listed options.
The CGenFF assigned parameters, by default, are output in CHARMM compatible format. GROMACS compatible topology and parameter files can be specified by selecting the “GROMACS Format” option.
If your input Mol2 does not have unique atom names, the CGenFF program will rename the atoms and the parameter file will reflect the names of the renamed atoms. In this case, it is advised to select the “Output Mol2” option.
In the event that the input molecule results in an error, you may select the “Include Debug Info” to identify possible erroneous entries in the input Mol2 file (e.g., missing hydrogen atoms).
By default, the CGenFF program will only output newly generated parameters as users are expected to use the force field in conjunction with the CHARMM36 force field. The parameters for which CGenFF directly applies to the input molecule can be explicitly listed in the output parameter file by selecting the “Copy Existing Param” option.
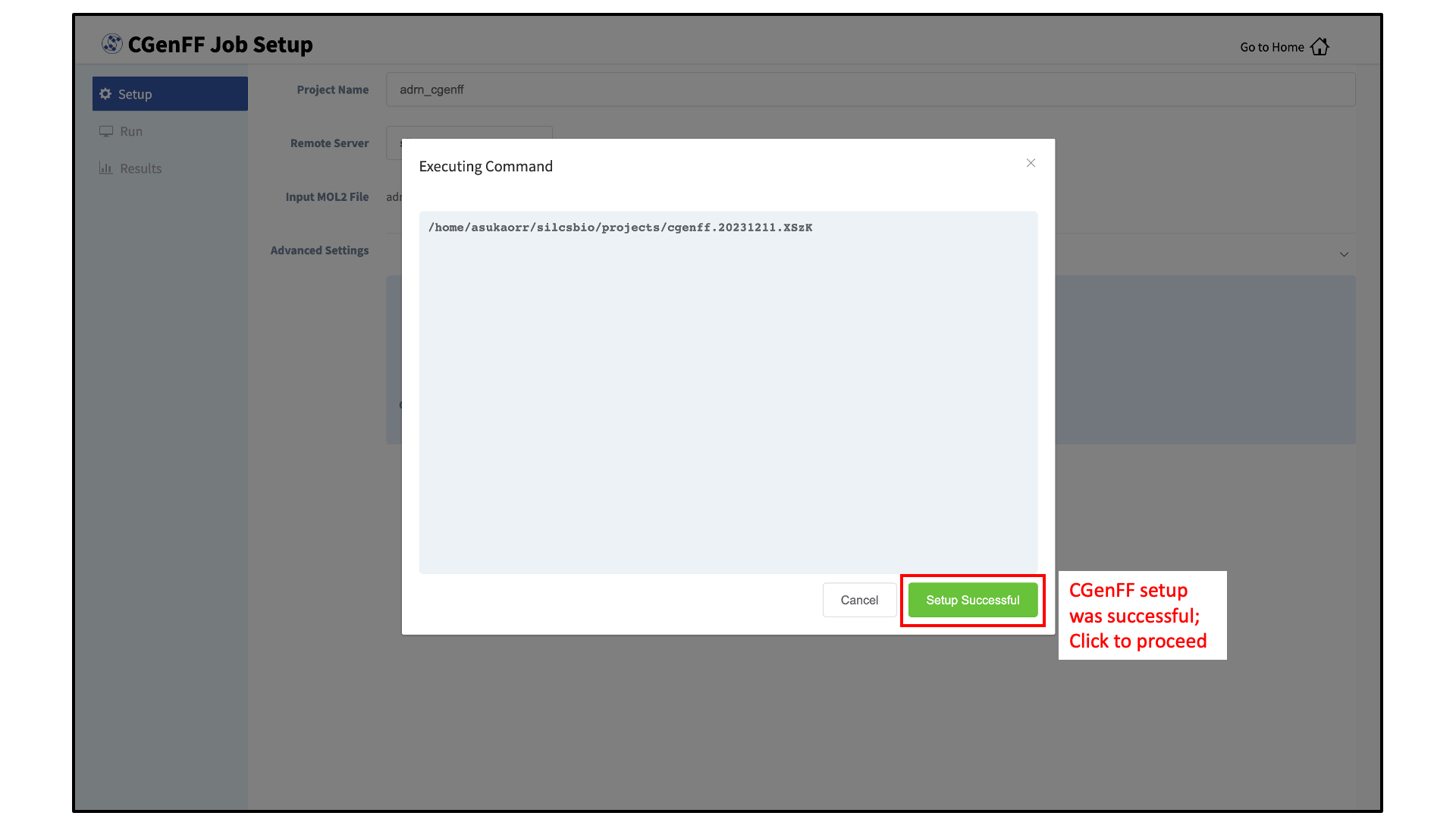
After all information is specified, click the “Setup” button to proceed. Once the CGenFF job is set up, the GUI will indicate that the setup was successful by displaying a green “Setup Successful” button. Click the green “Setup Successful” button to continue.
Review the ligand:
After setting up the CGenFF run, the GUI will prompt you to review your molecule. Click the “Review Molecule” button at the bottom of the screen to visualize the molecule. After reviewing the molecule, click “Run” in the sidebar menu to continue.
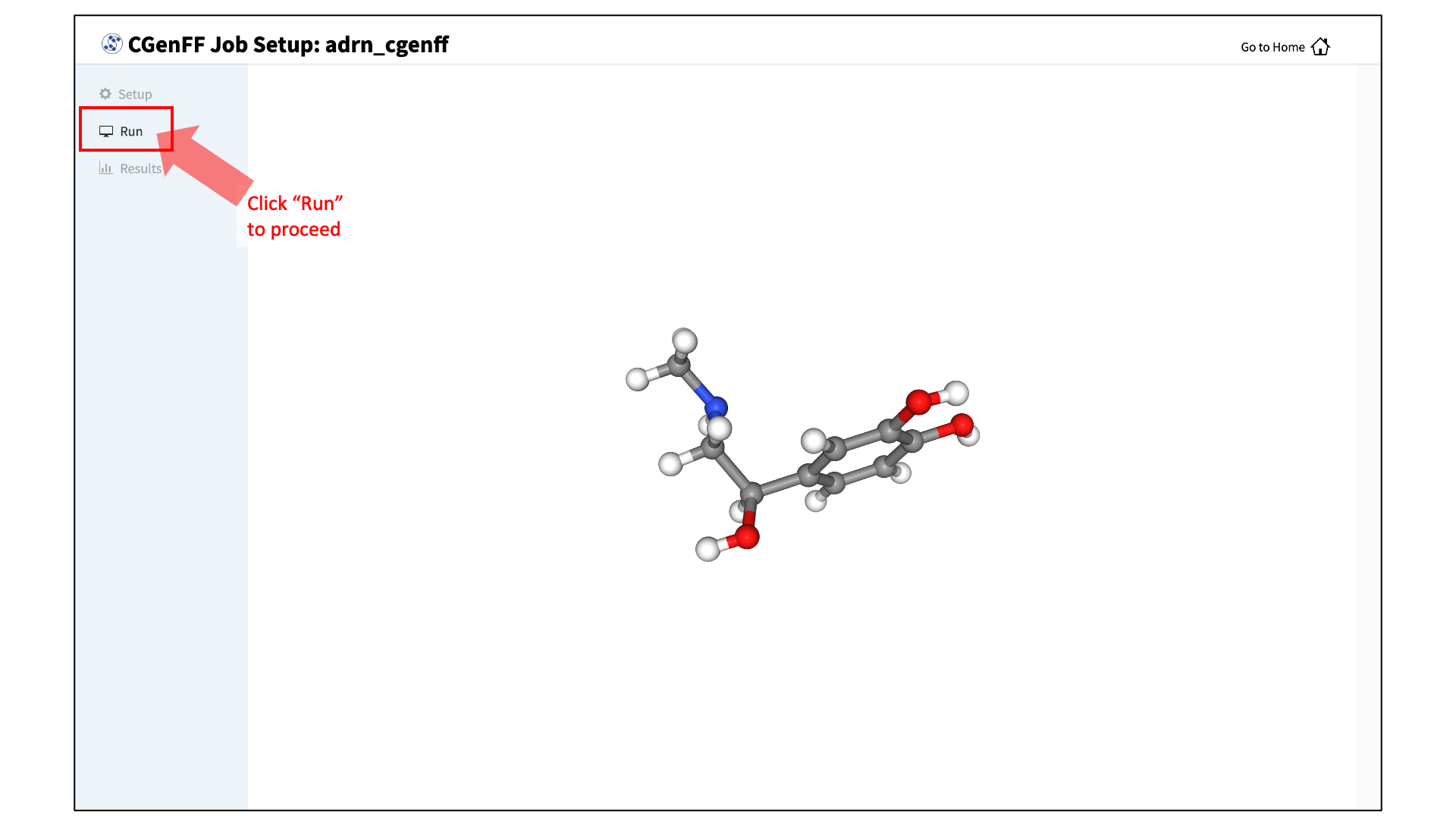
Launch the CGenFF job:
To run the CGenFF program, click the “Run CGenFF” button at the bottom of the page. You may also review the molecule again using the “Review Molecule Again” button.
Download CGenFF assigned topology and parameters:
Once the CGenFF program assigns force field parameters to the molecule, the results will be downloadable by clicking the green “Download Result” button.
For more information on the output stream file format and advanced CGenFF usage using CLI, please continue on to CGenFF Using the CLI. Please contact support@silcsbio.com if you need additional assistance.
CGenFF Using the CLI¶
The CGenFF program accepts Mol2 format files. For correct atom typing, it is important that all hydrogens are present, the system has correct protonation and tautomeric states, and that the bond orders are correct.
Once the Mol2 file is ready, the CGenFF program can be executed:
${SILCSBIODIR}/programs/cgenff <mol2file>
This produces a stream file in standard output. The output can be redirected to
a .str file. The stream file consists of a topology file and the extra
parameter file combined in a single output. It is up to the user to determine whether to
use the stream file directly or to split the file into two separate files.
As an example, the adrenalin molecule is parametrized in CGenFF. The example
input and output files can be found in
${SILCSBIO}/examples/cgenff/adrenalin.mol2 and
${SILCSBIO}/examples/cgenff/adrenalin.str, respectively.
The output stream file has two major sections.
* Toppar stream file generated by
* CHARMM General Force Field (CGenFF) program version 3.0
* For use with CGenFF version 4.6
*
read rtf card append
... topology section ...
END
read param card flex append
... parameter section ...
END
RETURN
In CHARMM, asterisk and exclamation marks represents comments. If you wish to
separate topology and parameter sections, you can take out the corresponding
section after read rtf or read param to the END statement.
Let’s take a look at topology section:
RESI ADRN 0.000 ! param penalty= 113.000 ; charge penalty= 16.607
GROUP ! CHARGE CH_PENALTY
ATOM O1 OG311 -0.649 ! 11.757
ATOM O2 OG311 -0.530 ! 0.000
ATOM O3 OG311 -0.530 ! 0.000
ATOM N NG311 -0.524 ! 16.607
ATOM C1 CG311 0.127 ! 16.171
Above is the beginning of the topology section. This section defines the atom names and types, as well as the atomic charges. Each entry is followed by a charge penalty score. The atomic partial charges are assigned based on chemical analogy to existing parametrizations in the force field. The penalty score becomes high when a good analogy is not found.
Below is an excerpt from the parameter section. When an exact parameter is not found in an existing parameter database, an entry is added based on an analogous parameter. Similar to atomic partial charges, each entry in the parameter section is also followed by a penalty score.
ANGLES
CG2R61 CG311 OG311 75.70 110.10 ! ADRN , from CG2R61 CG321 OG311, penalty= 4
CG311 CG321 NG311 43.70 112.20 ! ADRN , from CG331 CG321 NG311, penalty= 1.5
CG321 NG311 CG331 40.50 112.20 5.00 2.42170 ! ADRN , from CG3AM1 NG311 CG3AM1, penalty= 35
High penalty scores indicate that a parameter may be targeted for further optimization. Typically, penalty scores greater than 50 warrant further optimization.
It is possible to request that the CGenFF program preserve atomic partial
charges from the input Mol2 file instead of assigning these values. This is
done using the -z flag:
${SILCSBIODIR}/programs/cgenff -z <mol2file>
Warning
We STRONGLY advise against using your own custom atomic partial charges.
Nonbonded interactions in the force field depend upon both the Coulomb and
the Lennard-Jones terms, and the parameter values for these terms have been
carefully developed together. Assigning custom atomic partial charges
with the -z flag is likely to disrupt the balance in these nonbonded
force field terms.
Additional options¶
-hor--help: Displays help message and exits.--version: Displays version and licensing information, then exits.-qor--quiet: Suppresses all non-fatal warnings.-vor--verbose: Displays more warning messages. Use twice to also include debug information.-for--output-file(filename): Default: - (stdout)-mor--message-file(filename): Determines where to write warning, error and debug messages. Default: - (errstr)-aor--all-parameters: Copies existing parameters to the output. This is of very limited use because the atom types and L-J are still missing.-ior--input-format(format): Determines format for input molecule. Currently, only the default (Mol2) format is supported, so this flag is of no use.-wor--output-mol: Outputs processed Mol2 file of the molecule after assigning parameters.-xor--split-output(prefix): splits the output into separate .rtf and .prm files along with the unified .str file
More Advanced options¶
-bor--bond-order: Discards input bond orders and forces assignment of bond orders based on the connectivity instead. This may NOT be useful for 3rd-row and heavier elements.-eor--force-exhaustive: Forces using “exhaustive algorithm” for finding new parameters. This is usually slower but might be faster in some situations.-sor--server: Makes the program run in “server mode”, ie. a separate, self-contained output is generated for each input molecule.-por--parameter-file: Reads in parameter files. Ordinarily, parameter files should be read from the rule file.-lor--short-lone-pair: Put halogen lonepair particles closer to the atom-zor--keep-mol2-charge: Keep charge values from Mol2 file
GROMACS-readable parameters¶
To get parameters in GROMACS format, execute the following command.
${SILCSBIODIR}/cgenff/cgenff_to_gmx mol=<mol2file>
This command produces three files <mol2file>_gmx.top, <mol2file>_gmx.pdb & <mol2file>_charmm.str.
<mol2file>_gmx.top/pdb contain the GROMACS readable parameters and the coordinate information.
<mol2file>_charmm.str contain CHARMM readable parameters.
Additionally, charmm36.ff directory containing the full CHARMM36 force field
is given as output. This directory can be directly used to run simulations in
GROMACS. For the ligand with filename ligand.mol2, the charmm36.ff
directory will contain the following additional files:
lig_ffbonded.itp: Contains bonded parameters for the ligandlig.rtp: Contains the residue topology for the ligand