CGenFF for Covalent Ligands¶
The CGenFF program can be used to parameterize modified amino acids covalently bound to ligands of interest. Currently, users can covalently link ligands to cysteine (CYS), lysine (LYS), tyrosine (TYR), serine (SER), threoine (THR), asparagine (ASN), and glutamate (GLU) amino acids. The resulting parameters, in CHARMM and GROMACS compatible formats, can be used to simulate a protein with a covalently bound ligand.
CGenFF for Covalent Ligands Using the CLI¶
SilcsBio provides the utility to assign topology and parameters to modified amino acids covalently bound to ligands of interest only in the command line interface (CLI). The following steps describe how to generate topology and parameters for modified amino acids as well as build protein structures mutated with the modified amino acids covalently bound to the ligand of interest.
Prepare the ligand Mol2:
The CGenFF program accepts Mol2 format files. For correct atom typing, it is important that the structure meets the following criteria:
all hydrogens are present
protonation and tautomeric state is correct
bond orders are correct
The state of the ligand in the covalent bond should be considered when preparing the ligand Mol2 file. For example, the C=C double bond of an acrylamide molecule should be changed to a C-C single bond for a covalently bonded acrylamide molecule. Additionally, the ligand must be docked into its binding pocket in the desired orientation with respect to the amino acid to which it will be covalently linked. If the user has a current SILCS-Small Molecule Suite license, then this may be accomplished using SILCS-MC docking, described in SILCS-MC Docking Using the CLI or SILCS-MC Docking Using the SilcsBio GUI of SILCS-MC: Docking and Pose Refinement, with the target residue selection corresponding to the covalently bound amino acid.
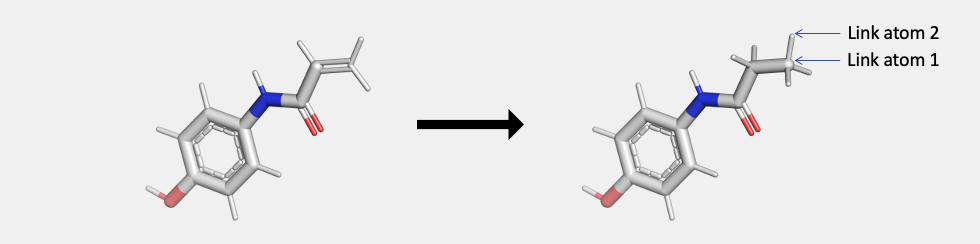
Once the Mol2 file is ready, note the atom ID for two atoms, Link atom 1 and Link atom 2 before you proceed to next step. Link atom 1 is the parent atom that will form a covalent bond with the amino acid side chain. Link atom 2 is the child atom that will get deleted once the bond is formed.
Warning
The ligand Mol2 file must contain coordinates of the ligand within the desired binding pocket and in the proper orientation with respect to the amino acid to which the covalent bond will be formed. This can be conveniently acheived using SILCS-MC docking, described in SILCS-MC Docking Using the CLI or SILCS-MC Docking Using the SilcsBio GUI of SILCS-MC: Docking and Pose Refinement, with the target residue selection corresponding to the covalently bound amino acid.
Build the modified amino acid covalently bound to the ligand:
Using the chemical group transformation module available through the SilcsBio software (Chemical Group Transformations), the ligand will be covalently linked to a desired amino acid (cysteine (CYS), lysine (LYS), tyrosine (TYR), serine (SER), threoine (THR), asparagine (ASN), or glutamate (GLU)). A new Mol2 file will be generated containing the energetically minimized, covalently bound amino acid.
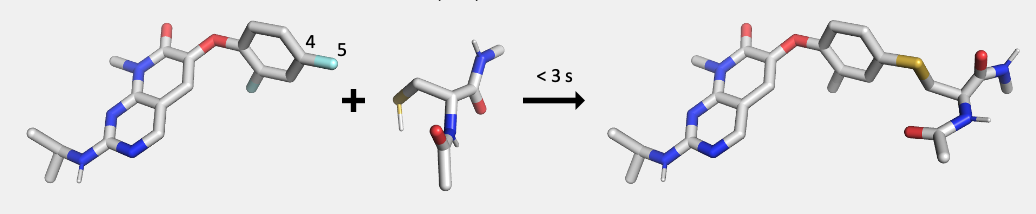
Use the following command to generate the structure of the input ligand covalently bound to the desired amino acid:
$SILCSBIODIR/cgenff-covalent/1_build_mod_res lig=<ligand mol2 file> linkatom1=<parent atom ID> linkatom2=<child atom ID> linkresname=<linking amino acid>Required parameters:
The path and name of the input ligand Mol2 file:
lig=<location and name of ligand mol2>The atom ID (integer value) of the parent atom that will form the covalent bond with the amino acid side chain:
linkatom1=<atom ID for parent atom>The atom ID (integer value) of the child atom that will get deleted once the covalent bond to the amino acid side chain is formed.
linkatom2=<atom ID for child atom>Note
Please ensure that the atom IDs entered for
linkatom1andlinkatom2correspond to the input Mol2 file entered forlig. The atom ID is listed in the first column of the Mol2 file under the@<TRIPOS>ATOMsection.
The residue name of the bound amino acid for covalent bonding with the ligand. The SilcsBio software supports covalent bonds to be formed with cysteine (CYS), lysine (LYS), tyrosine (TYR), serine (SER), threoine (THR), asparagine (ASN), and glutamate (GLU):
linkresname=<linking amino acid; CYS/LYS/ASN/SER/THR/TYR/GLU>Optional parameter:
The residue name and the basename of the output Mol2 file of the covalently bound amino acid. By default,
CVLis used as the residue name:newresname=<new residue name; default=CVL>With the default
newresname,CVL, the output from this step will becvl.mol2.
Generate the topology and parameters of the covalently bound amino acid:
In this step, the CGenFF program generates the topology and parameters for the modified amino acid in Mol2 format created in the previous step. The topology and parameters for the modified amino acid are output in formats compatible with both the CHARMM and GROMACS MD simulation packages. The CHARMM compatible format will be output in a stream file (
.strfile). The GROMACS compatible format will be output in a directory (charmm36.ff).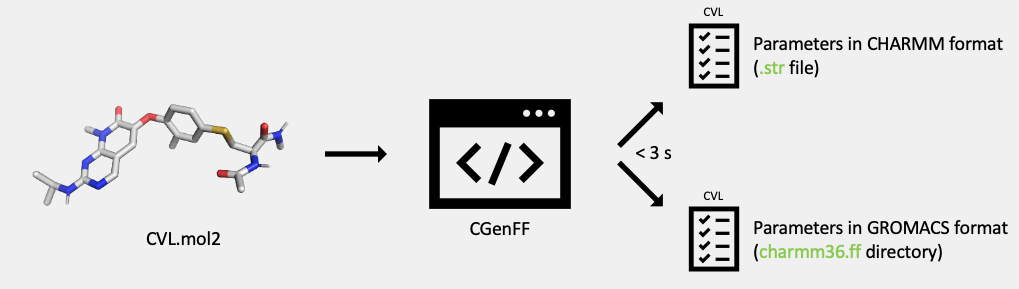
The topology and parameters of the modified, covalently bound amino acid is converted to the topology and parameters of a residue that can be incorporated into a full protein sequence. The atom types of the backbone atoms of the modified amino acid are converted from CGenFF atom types to CHARMM36 atom types compatible with the CHARMM36m protein force field.
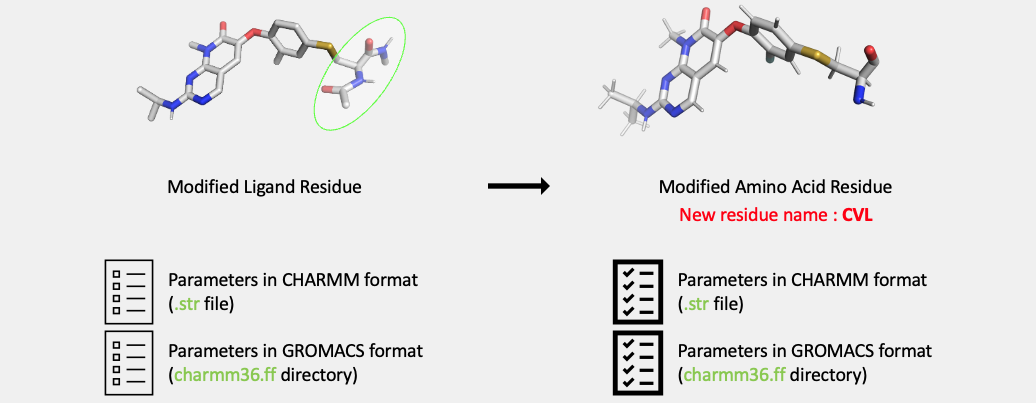
To generate the topology and parameters of the covalently bound amino acid, use the following command:
$SILCSBIODIR/cgenff-covalent/2_gen_rtp_pdb cvl=<covalently bound amino acid mol2> linkresname=<linking amino acid>Required parameters:
The path and name of the covalently bound amino acid Mol2 file. This file corresponds to the output Mol2 file from the previous step:
cvl=<location and name of the covalently bound amino acid mol2>The residue name of the amino acid to which the ligand molecule is covalently bound. This entry corresponds to the same
linkresnameused in the previous step:linkresname=<linking amino acid; CYS/LYS/ASN/SER/THR/TYR/GLU>Optional parameters:
The residue number (integer value) of the covalently bound amino acid. The residue number of the covalently bound amino acid may be set to its corresponding residue number in the protein to which the ligand is covalently bound. This value can additionally be overwritten in the next step. The default value is set to
1:linkresid=<linking amino acid resid; default=1>The chain ID of the covalently bound amino acid. The chain ID of the covalently bound amino acid can be defined in accordance with the target protein PDB:
linkchain=<linking amino acid chain ID; default=NULL>With the default
newresname,CVL, the outputs from this step will include the following:
cvl.pdb
cvl.rtp
toppar_cvl.str
charmm36.ff
toppar_all36_prot_modify_res.str
Mutate the protein:
The protein structure can now be modified to incorporate the covalently bound amino acid. In this step, the specified amino acid is mutated to the desired covalently bound amino acid in the input target protein PDB file. The structure, topology, and parameters of the covalently bound amino acid correspond to those generated in the previous steps.
Use the following command:
$SILCSBIODIR/cgenff-covalent/3_mutate_to_cvl prot=<protein PDB file> cvl=<CVL PDB file> linkresname=<linking amino acid> linkresid=<mutated residue ID>Required parameters:
The path and name of the target protein PDB file:
prot=<location and name of protein PDB file>The path and name of the covalently bound amino acid PDB file. This file corresponds to the output PDB file from the previous step:
cvl=<location and name of the covalently bound amino acid pdb>The residue name of the amino acid to which the ligand molecule is covalently bound. This entry corresponds to the same
linkresnameused in the previous steps:linkresname=<linking amino acid; CYS/LYS/ASN/SER/THR/TYR/GLU>The residue number (integer value) of the covalently bound amino acid. The residue number of the covalently bound amino acid should be set to its corresponding residue number in the protein to which the ligand is covalently bound:
linkresid=<linking amino acid resid>Optional parameters:
The name of the output PDB file. The default name of the output pdb is set to
prot-cvl.pdb:outputpdb=<name of the output PDB file; default="prot-cvl.pdb">
Tip
You may wish to try the example provided in ${SILCSBIODIR}/examples/cgenff-covalent/
to familiarize yourself with the CGenFF for Covalent Ligands workflow.
The outputs of the CGenFF for Covalent Ligands workflow may be used for Standard Molecular Dynamics (MD) Simulations or,
with the SILCS-Small Molecule Suite, SILCS Simulations. For these simulations,
the initial structure will correspond with the outputpdb,
prot-cvl.pdb by default.
To perform standard MD simulations of a protein with a covalent ligand, follow the instructions provided in Standard Molecular Dynamics (MD) Simulations with
covalent=truespecified for the first,${SILCSBIODIR}/md/1_setup_md_boxesstep. Specify theoutputpdbfrom this section (prot-cvl.pdbby default) as the initial structure withprot=prot-cvl.pdb.To perform SILCS simulations of a protein with a covalent ligand, the user must have a current license for the SILCS-Small Molecule Suite. Please contact info@silcsbio.com for more information. To set up the SILCS simulations, follow the instructions provided in SILCS Simulations Using the CLI. In the first,
${SILCSBIODIR}/silcs/1_setup_silcs_boxesstep, specify theoutputpdbfrom this section (prot-cvl.pdbby default) as the initial structure withprot=prot-cvl.pdb. This will produce an error due to theCVLresidue being unrecognized. In the resulting1_setupdirectory, replace thecharmm36.ffdirectory with the GROMACS formatted output from CGenFF-covalent, also namedcharmm36.ff. Continue the setup process by repeating the first,${SILCSBIODIR}/silcs/1_setup_silcs_boxesstep withskip_pdb2gmx=true. Continue to the following steps as described in SILCS Simulations Using the CLI. Note that it is typically NOT recommended to run a SILCS simulation with any ligand bound in the binding pocket.
For simulations with the CHARMM program, the outputpdb, prot-cvl.pdb by default, will
need to be edited to fit CHARMM PDB format (e.g., adding the segname column) and
the output topology and parameter stream files, toppar_cvl.str and
toppar_all36_prot_modify_res.str by default, should be added to the CHARMM
input.