Background¶
Biomacromolecular therapeutics, commonly called “biologics,” are typically protein molecules that have been developed to selectively interact with a therapeutic target. Common examples of biologics are therapeutic antibodies. Biologics must be carefully formulated to maximize their stability, so as to ensure both efficacy and safety. Important determinants of stability are protein aggregation and protein denaturation. Toward maximizing stability, biologics can be formulated with excipients, which can help minimize aggregation and denaturation of the biologic in a solution formulation.
SILCS-Biologics applies the patented Site Identification by Ligand Competitive Saturation (SILCS) platform technology to the rational selection of excipients for biologics formulations. The first goal is to minimize protein–protein interactions between multiple molecules of the protein, all in the native state. This reduces the likelihood of aggregation of the protein in its native state. The second goal is to minimize denaturation by stabilizing the native (folded) state of the protein. This is accomplished by screening for excipients that can efficiently bind to the protein native state. Such binding drives the equilibrium toward the native state and away from denatured states. Achieving the second goal also contributes to minimizing aggregation, as denatured states of the protein can be aggregation-prone.
The SILCS-Biologics workflow consists of four steps: 1) SILCS simulation, 2) protein–protein interaction screening, 3) protein-excipient hotspot screening, and 4) analysis and ranking.
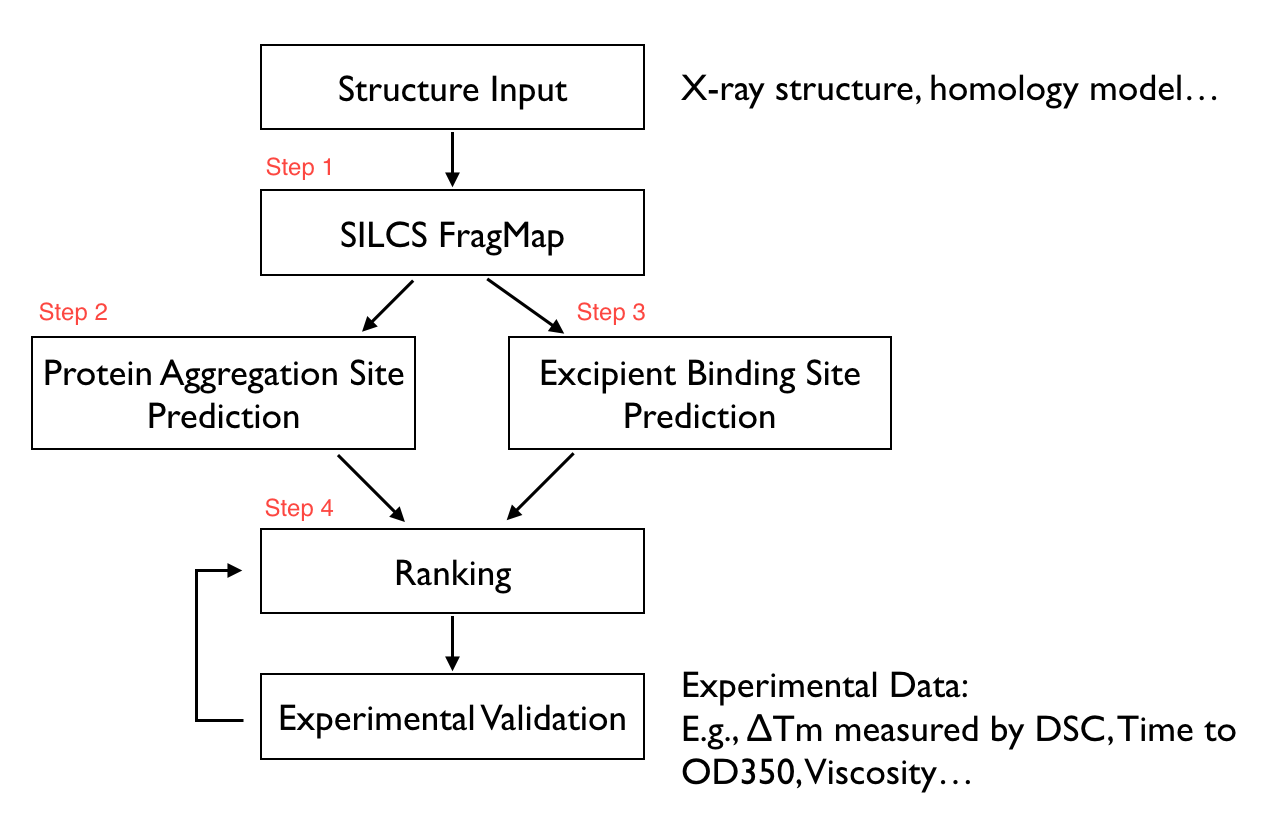
SILCS-Biologics is actualized as a unified tool, available through both the command line and the SilcsBio Graphical User Interface, that integrates all four steps. As such, SILCS-Biologics automatically takes care of preparing and running computing jobs that entail SILCS to generate FragMaps, SILCS-PPI to predict protein–protein interactions (PPI) that can drive aggregation, and SILCS-Hotspots to determine excipient binding sites. Please see SILCS Simulations and SILCS-Hotspots for additional details. SILCS-PPI works by aligning FragMaps from one protein with the functional groups on the surface of another protein to predict the likelihood of a PPI between the two proteins [1]. Please see Protein–Protein Interaction Maps for additional information.