SILCS-Hotspots¶
Background¶
SILCS-Hotspots identifies all potential fragment binding sites on a target by applying SILCS-MC sampling on fragment-like molecules across the entire target structure. Refer to SILCS-MC Docking Protocol Details for technical details on SILCS-MC sampling of fragment molecules in translational, rotational, and torsional space in the field of the FragMaps. The protein or other macromolecular target is partitioned into a collection of subspaces in which the fragment is randomly positioned and subjected to extensive SILCS-MC to identify favored local poses. This may be performed 1000 times or more per fragment in each subspace. The identified fragment positions are then RMSD-clustered followed by selection of the lowest energy pose in each cluster to define a binding site for further analysis.
SILCS-Hotspots may be run using small fragment-like molecules as well as larger drug-like molecules. When multiple fragments are used, a second round of clustering may be performed over the different fragment types to identify binding sites occuppied by one or more of the fragment types included in the calculation.
Results from SILCS-Hotspots include all potential binding poses of the individual fragments, binding sites that contain one or more of the fragment types, and ranking of fragment poses and binding sites based on LGFE scores. Applications of SILCS-Hotspots include identifying putative fragment binding sites and performing fragment-based drug design. Putative binding sites identified by SILCS-Hotspots can be used for rapid database screening with SILCS-Pharm. Alternatively, sites identified by SILCS-Hotspots can be considered for fragment-based design by linking poses of fragment-like molecules in adjacent sites to create drug-like molecules.
Inputs for SILCS-Hotspots are the protein or other macromolecular target used for the SILCS simulations, the SILCS FragMaps resulting from those simulations, and the fragment(s) to be used for sampling. SilcsBio provides fragment files appropriate for SILCS-Hotspots (see below). Alternatively, you may use your own fragment file(s).
SILCS-Hotspots Using the SilcsBio GUI¶
Begin a new SILCS-Hotspots project:
Select New SILCS-Hotspots project from the Home page.
Enter a project name and select the remote server:
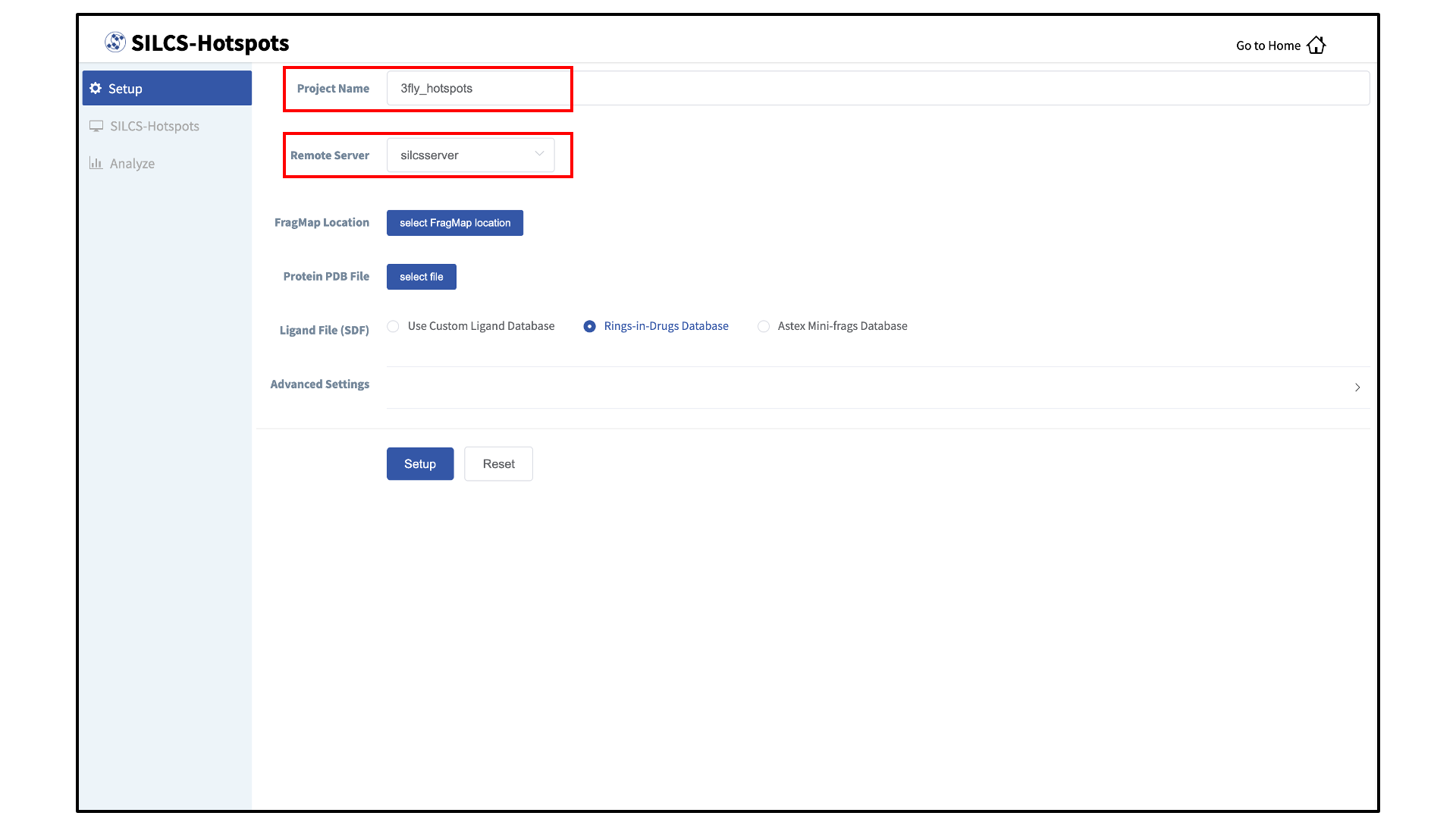
Enter FragMap, protein, and ligand input files:
Provide FragMap and protein input PDB files. You will additionally need to provide a “Ligand file (in SDF format)” that contains the database of ligands to be used for sampling (see Default databases of fragment-like molecules). You may choose these files from your local machine where you are running the SilcsBio GUI (“localhost”) or from any server you have previously configured, as described in File and Directory Selection.
Warning
Ligands in the “Ligand file (in SDF format)” must include all hydrogens, including pH-appropriate (de)protonations, and must have reasonable three dimensional conformations.
Upload input files to the server:
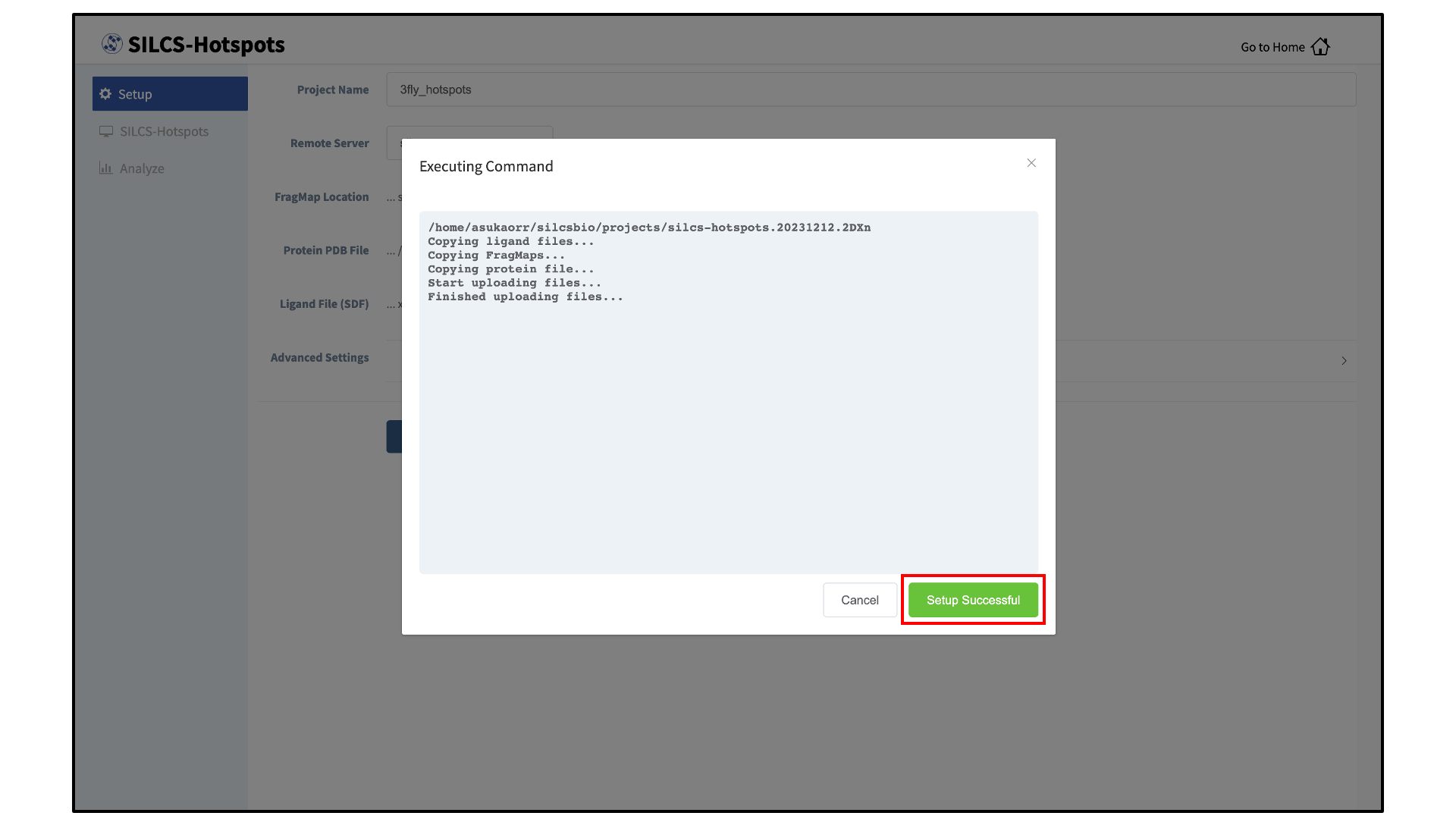
Once the information is entered correctly, click the “Setup” button at the bottom of the page. The GUI will contact the remote server and upload the input files to the “Project Location” directory on the remote server. A green “Setup Successful” button will appear once the upload has successfully completed. Press this green button to proceed.
Launch SILCS-Hotspots jobs:
The GUI will display a summary screen with the Project Name, Remote Server, Project Location, Protein PDB file, Ligand SDF file, and the Number of Ligands. You will also see “Sampling Mode” and “Bundle” under “Advanced Settings”. You may select/adjust these if you desire. Click on the “Run SILCS-Hotspots” button. Doing so will submit jobs to the remote server and list them in a pop-over window.
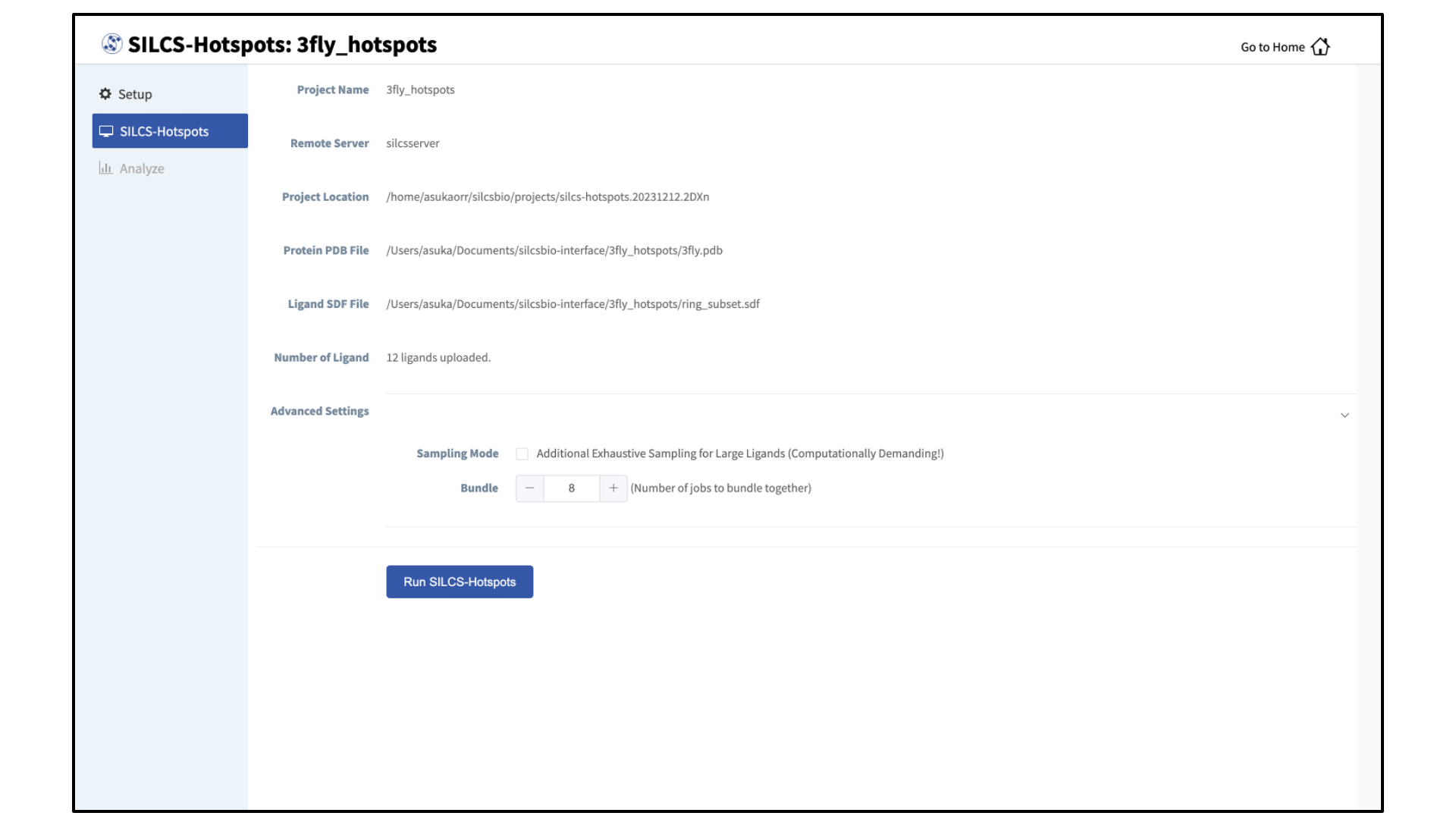
Once the jobs are submitted, you may click on the “Setup Successful” button to dismiss the pop-over window and return to the previous screen.
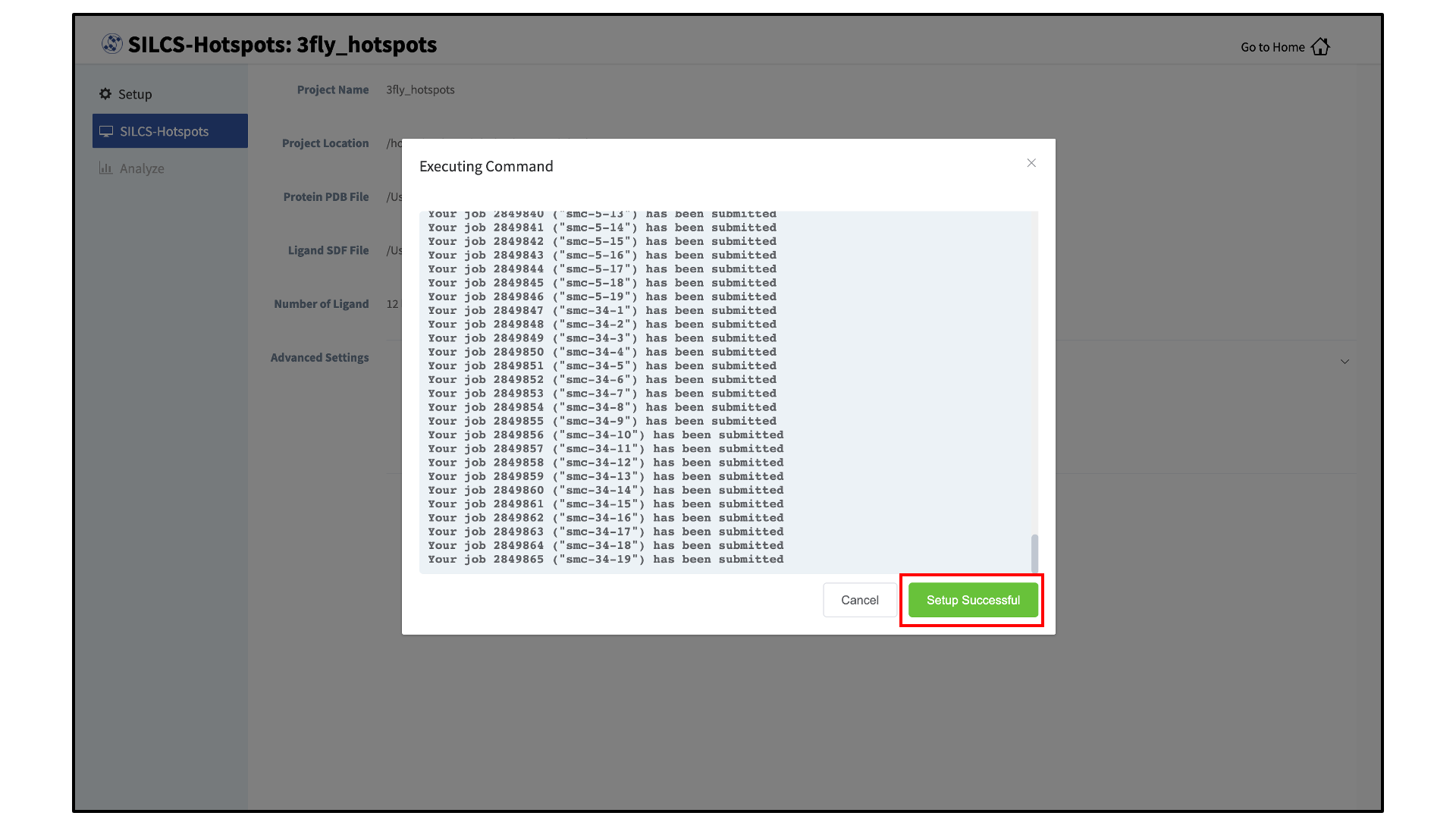
Monitor SILCS-Hotspots jobs:
The screen will now show a “Simulation Progress” section. You can update this section by clicking the “Refresh” button. This will update the job progress bars in the SilcsBio GUI.
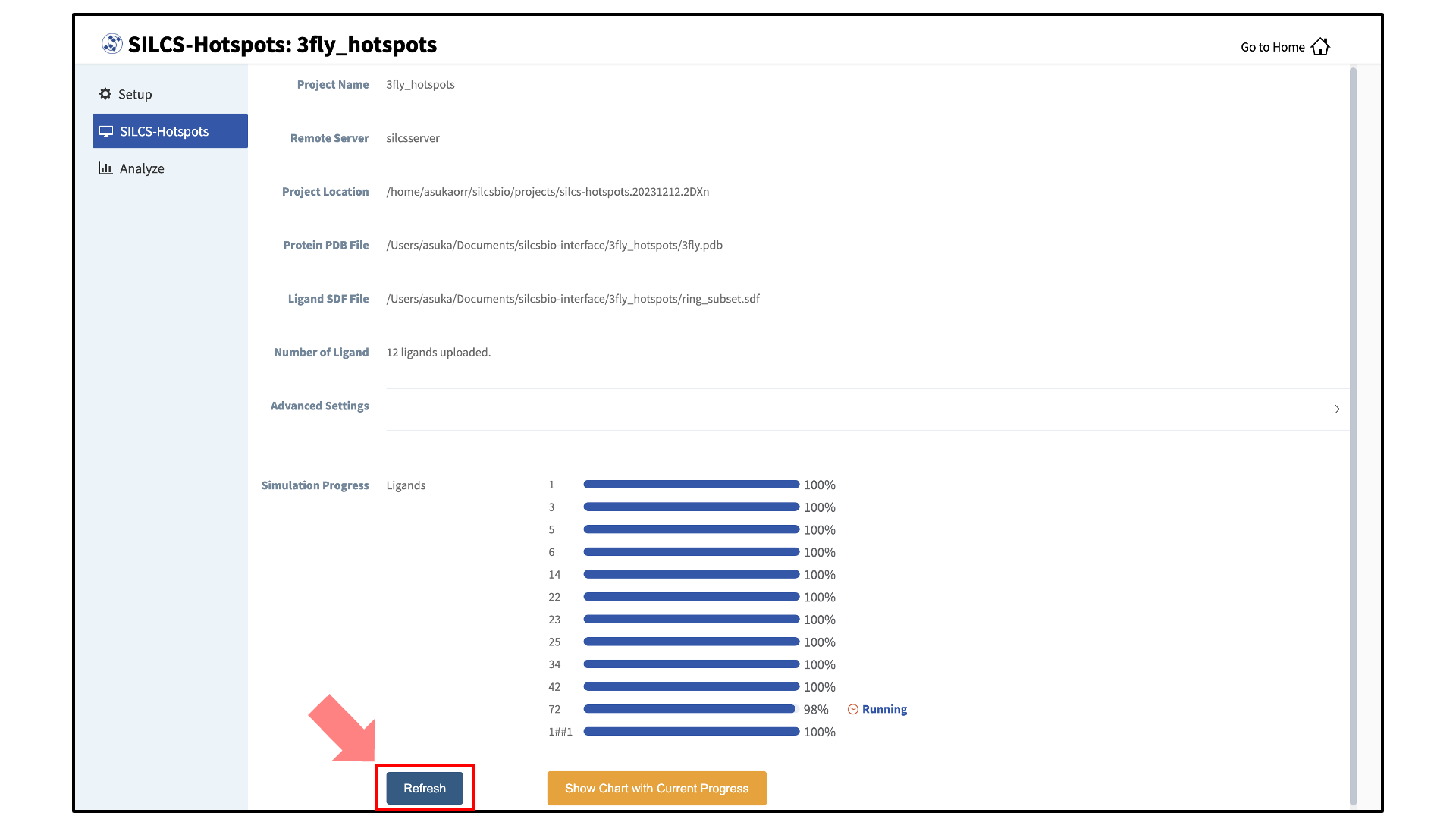
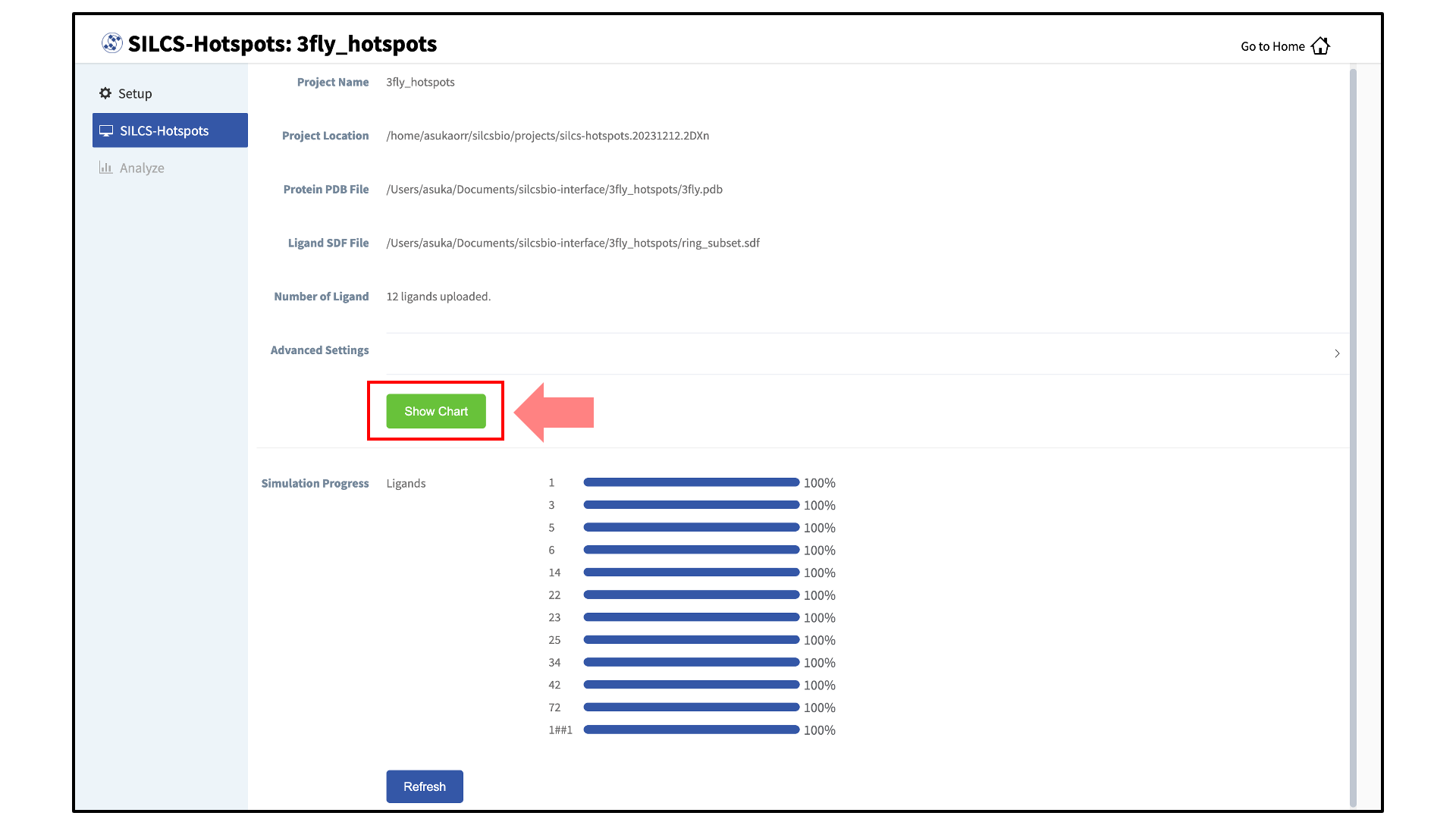
Once the progress bars reach 100%, you will see a green “Show Chart” button. Click it to proceed. You may also click on the “Show Chart with Current Progress” button while some of the jobs are still running.
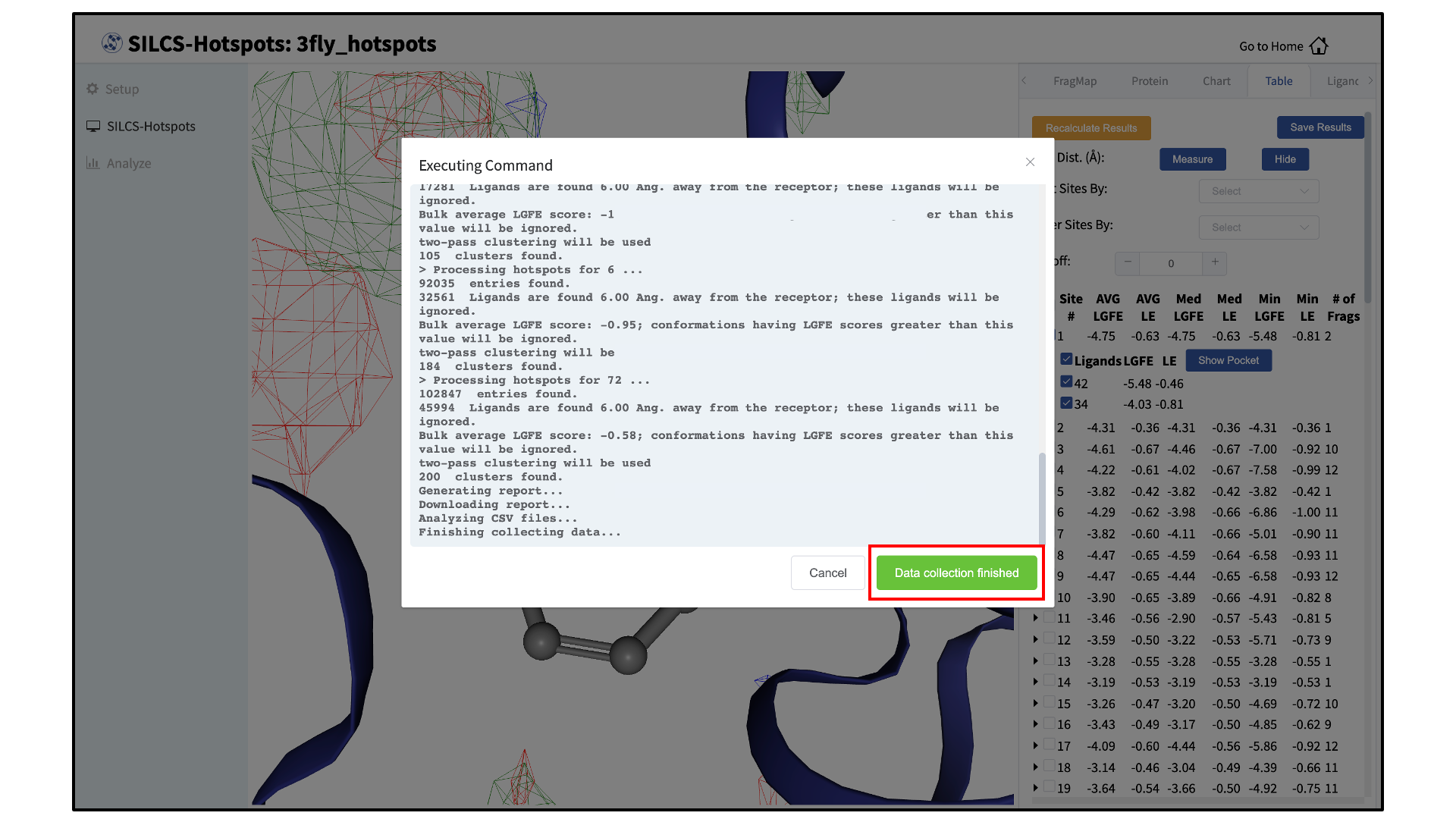
Collect SILCS-Hotspots data:
Clicking the green “Show Chart” button will download a report folder from the server. The GUI will automatically update once all of the necessary data is collected. Click on “Data collection finished” to proceed.
Note
If you encounter an error in this step, it is likely that required Python 3 packages have not been installed. Please refer to Python 3 Requirement for more information.
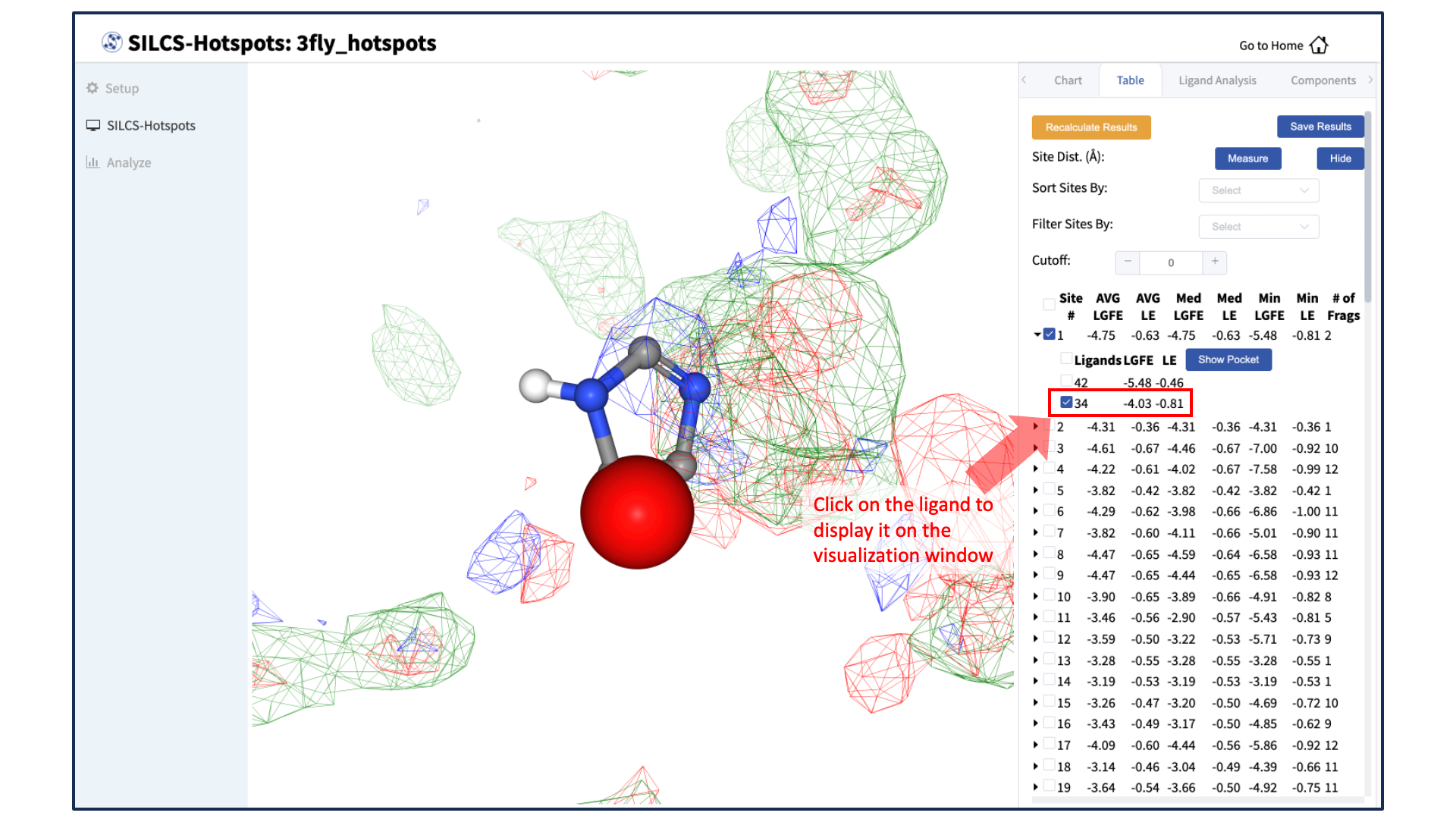
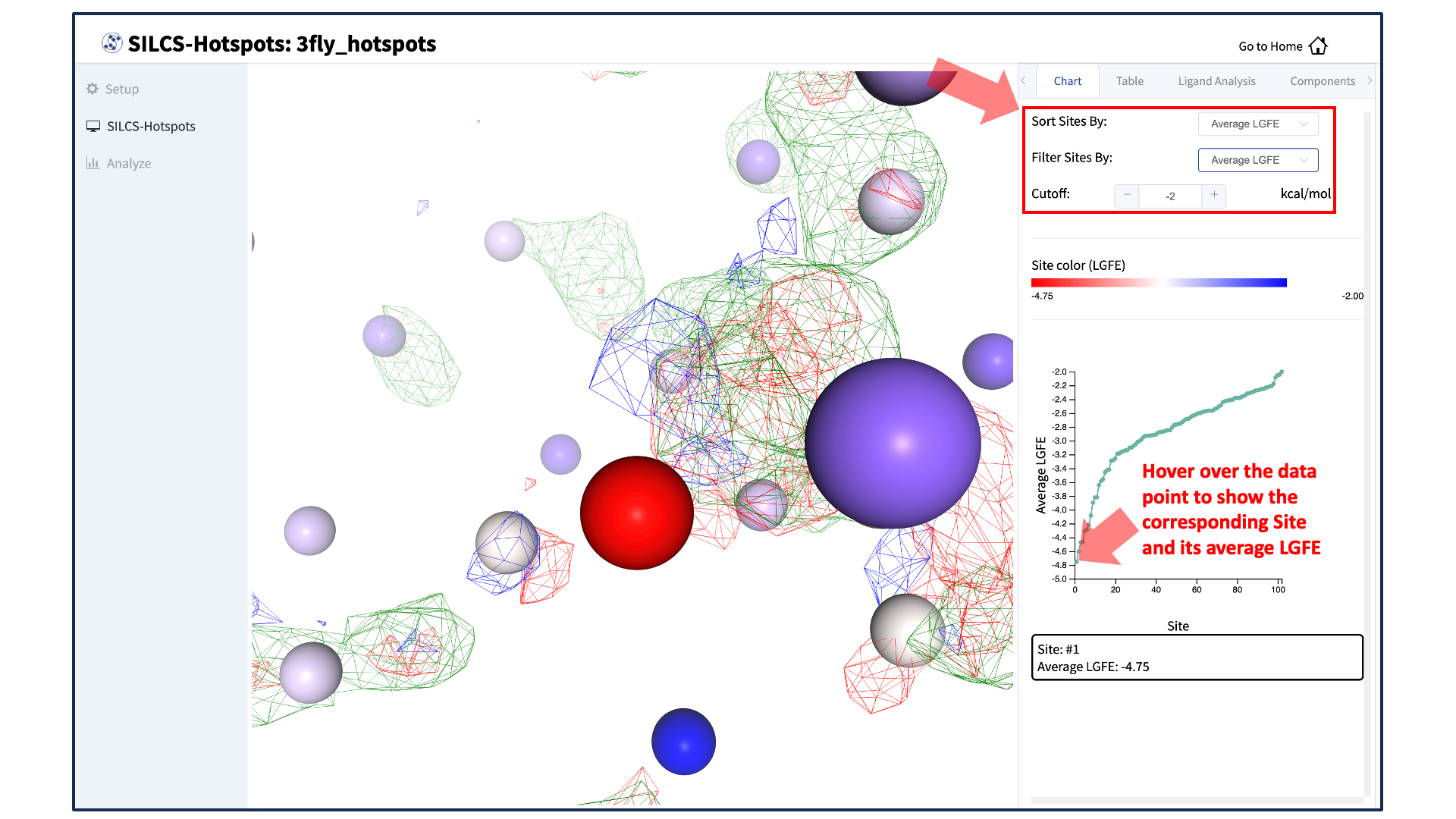
Visualize SILCS-Hotspots results:
A new tab, labeled “Table” will have been created in the right-hand panel. The “Table” tab lists the LGFE and Ligand Efficiency (LE) scores of each hotspot (referred to as “site”). By clicking on the ligand under the “Table” tab, you can center the visualization window at the ligand.
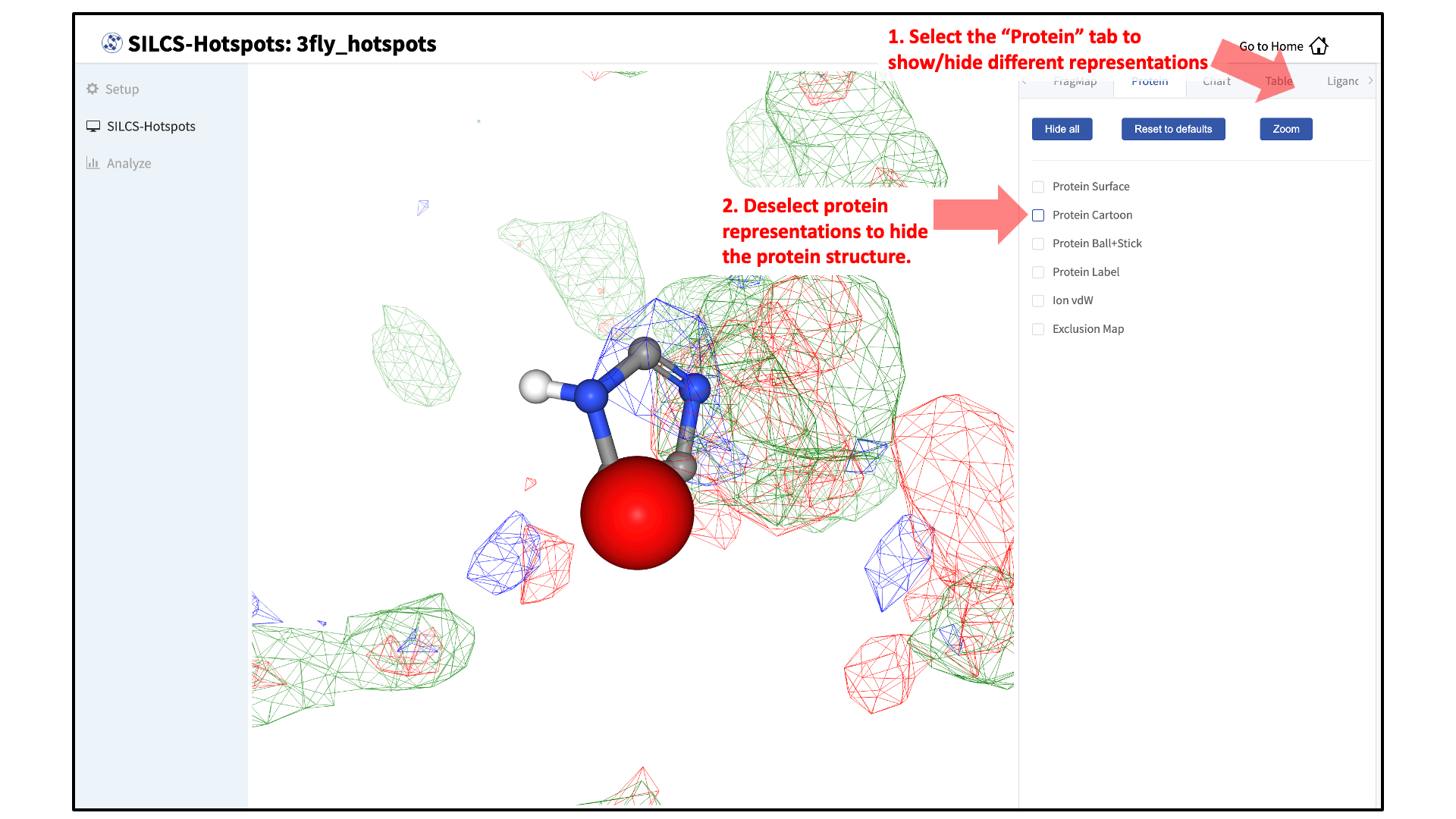
Customize hidden/shown components:
You may also click on the “Components” tab in the right-hand panel and deselect the protein for easier viewing.
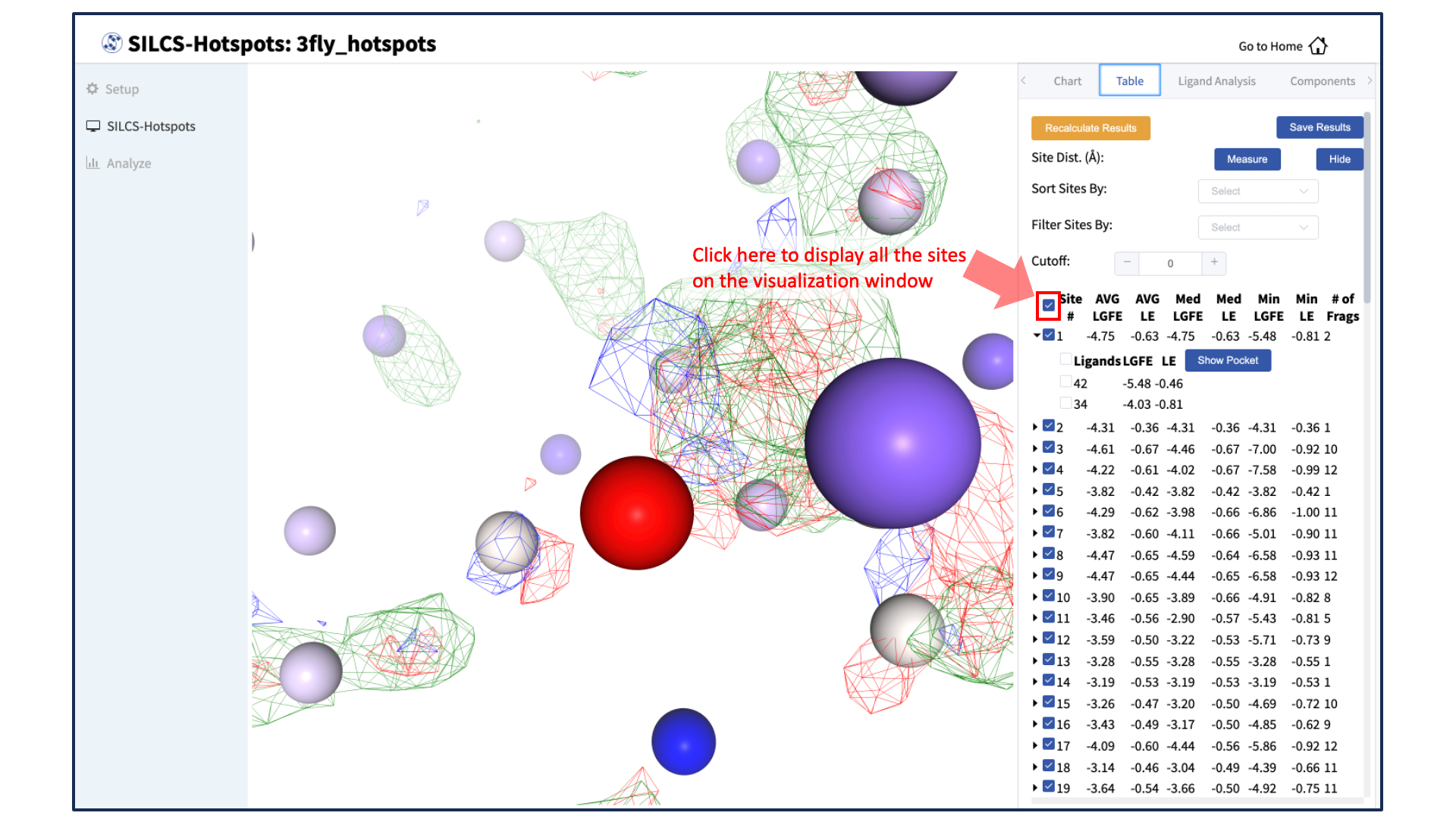
Save SILCS-Hotspots docked structures and data to local computer:
Click on the box next to “Site #” to display all the hotspot sites in the visualization window. Click the “Save Results” button to save the SILCS-Hotspots results for all your selected hotspot sites to your local computer. You can sort the table using the “Sort Site By:” drop-down menu. You can additionally display only those sites meeting a cutoff criterion by using the “Filter Sites By:” drop-down menu and adjusting the “Cutoff:” value.
Measure distances between hotspot sites:
To measure the distance between two hotspot sites, select their solid spheres and click the “Measure” button in the right-hand panel. This will display a dashed line in the visualization window with the measured distance (in Å) from one hotspot site to the other.

Plot average LGFE of SILCS-Hotspots sites:
The “Chart” tab in the right-hand panel converts the information from the “Table” tab into plots. The plot can be adjusted by sorting and/or filtering sites using the “Sort Sites By:” and “Filter Sites By:” drop-down menus. Changing the “Cutoff:” value will adjust the y-axis. The plot is interactive: hovering your mouse over a point will display the x and y values for that point.
SILCS-Hotspots Using the CLI¶
Launch SILCS-Hotspots SILCS-MC jobs:
Input arguments for launching the SILCS-Hotspots SILCS-MC jobs are: the PDB file used for the SILCS run (
prot), the location of the fragment files (ligdir), and the location of the SILCS FragMaps (mapsdir). When SILCS-Hotspots calculations are performed, the subdirectory4_hotspots/is created to store output from the exhaustive SILCS-MC runs.$SILCSBIODIR/silcs-hotspots/1_setup_silcs_hotspots prot=<prot PDB> ligdir=<fragment database> mapsdir=<mapsdir> bundle=<true/false>
Note
The above command will submit a large number of jobs, which can strain job queueing systems. The
bundlekeyword launches bundled jobs instead of individual jobs.Required parameters:
Path and name of the input protein PDB file:
prot=<protein PDB file>
Fragment database location:
ligdir=<location and name of directory containing fragment mol2/sdf>
See Default databases of fragment-like molecules for detailed information.
Note
.sdf,.sd, or.mol2files can be placed in theligdirdirectory, and SILCS-MC will read a single molecule from each file. If a file contains multiple molecules, use of theligdiroption will result in only the first molecule in the file being processed.If you have an SDF file with multiple molecules in it, replace
ligdir=<directory containing ligand mol2/sdf>withsdfile=<path to sdfile>to process all molecules in the file.Warning
Ligands, regardless of file format, must include all hydrogens, including pH-appropriate (de)protonations, and must have reasonable three dimensional conformations.
Optional parameters:
FragMap directory path:
mapsdir=<location and name of directory containing FragMaps; default=maps>
By default, the program looks for FragMaps in the
maps/directory.Option to bundle jobs:
bundle=<true/false; default=true>
Note
The
1_setup_silcs_hotspotscommand will submit a large number of jobs, which can strain job queueing systems. Running the command with thebundlekeyword will submit larger bundled jobs instead of many individual jobsNumber of jobs to bundle (when
bundle=true):nproc=<number of jobs to bundle; default=8>
Directory containing output to be used in subsequent steps:
hotspotsdir=<location of hotspots data; default=4_hotspots>
Template file for SILCS-MC sampling:
paramsfile=<custom params file>
The default SILCS-MC job parameters file for SILCS-Hotspots is
$SILCSBIODIR/templates/silcs-hotspots/params.tmpl. See User-Defined Protocols through the CLI for customization details.Location and name of optional SD file:
sdfile=<location and name of SD file>
Note
For SILCS-Hotspots jobs with many fragment ligands, an SD file containing all of the fragments rather than individual Mol2 files can be used.
Exhaustiveness of SILCS-MC sampling:
exhaustive=<true/false; run longer sampling for larger than fragment molecules; default=false>
Perform post-run clustering:
Once the SILCS-Hotspots SILCS-MC jobs are complete, the next step is to cluster fragment poses that were output by the completed jobs. The clustering algorithm iteratively finds clusters with the largest number of members [20]. The process entails 1) computing the number of neighbors of each pose, 2) choosing the pose with the largest number of neighbors and marking it and its neighbors as cluster members, 3) removing cluster members identified in Step 2 from the pool of poses, and 1) Steps 1-3 are automatically repeated until there are no available poses left. The cluster “center” is defined as that fragment pose with the most neighbors in that cluster. This pose’s LGFE score defines the LGFE score of the cluster. Run the command
$SILCSBIODIR/silcs-hotspots/2_collect_hotspots prot=<prot PDB> ligdir=<fragment database>
to perform the clustering. You may also specify the following additional options.
Optional parameters:
Radius for RMSD clustering:
cutoff=<cutoff for clustering; default=3>
Maximum number of sites to be identified for each fragment:
maxsites=<maximum number of sites ordered by LGFE; default=200>>
Directory containing output from the previous steps:
hotspotsdir=<location of hotspots data; default=4_hotspots>
Maximum allowable distance of the excipient from the protein
lig_max_dist=<maximum distance of ligands from the protein; default=6>
Location and name of optional SD file:
sdfile=<location and name of SD file>
Note
The SD file option should only be used if an SD file was used in the previous
1_setup_silcs_hotspotsstep. In this case, use the same SD file from the previous step in the post-run clustering,2_collect_hotspots, step.Determine binding site and generate summary report:
The final step of SILCS-Hotspots is site determination and report generation. This involves a second round of clustering over all specified fragments to identify sites on the protein to which one or more fragments bind. Each site is given the average LGFE score over all the fragments. In addition, the sites themselves may be clustered to identify binding pockets that suggest multiple adjacent sites that may be linked to build larger fragments (under development). Information on all the fragments, the binding sites, and the binding pockets is included in
report.xlsxand in PDB files that are output to the subdirectory4_hotspots/report/. Run the following command$SILCSBIODIR/silcs-hotspots/3_create_report ligdir=<fragment database>
Optional parameters:
Directory containing output from the previous steps:
hotspotsdir=<location of hotspots data; default=4_hotspots>
Cluster radius for binding site determination:
site_cutoff=<cluster radius for site determination; default=6.0>
LGFE cutoff for inclusion of fragment poses in site determination:
ligand_lgfe_cutoff=<Ligand LGFE cutoff for site determination; default=-2.0>
Average LGFE cutoff for site determination:
site_lgfe_cutoff=<average LGFE cutoff for binding sites; default=-2.0>
Sites having values less favorable than the cutoff will be discarded.
Flag to activate binding pocket calculation and associated clustering radius (under development):
pocket=<perform pocket analysis; default=False> pocket_cutoff=<cluster radius for binding pocket determination; default=12.0>
The
3_create_reportcommand will populate the4_hotspots/report/subdirectory with the following output files:report_all.xlsx: Spreadsheet file containing analysis information. Included are the LGFE and Ligand Efficiency (LE) scores for each copy of every fragment obtained from clustering, relative affinity analysis, and listing of the posed fragment that defines each site. If binding pocket analysis is performed, information on binding pockets is included.hotspots_sites.pdb: PDB file containing the identified sites. The B-factor column value includes the average LGFE score of that site.pdb_by_ligands: Directory containing PDB files for each fragment with multiple coordinates for each site identified for the fragment. In the PDB files, REMARK includes the LGFE scores, the B-factor column includes the GFE score for each atom, and the final column lists the SILCS atom type.pdb_by_site: Directory containing PDB files of the fragments located at each site inhotspots_sites.pdb. For example, the filenamesite_all_1_8_1.pdbindicates that at site 1 fragment 8 is present. The final 1 indicates that this is the first copy of fragment 8 at site 1. From the clustering alogrithm, it is possible that more than one copy of a fragment is included in a site. For example,site_all_1_8_2.pdbwould be the second copy of fragment 8. In each PDB file, REMARK includes the LGFE scores, the B-factor column includes the GFE score for each atom, and the final column lists the SILCS atom type.
The following additional outputs are produced if binding pocket analysis is performed (
pocket=true):hotspots_pockets.pdb: PDB file containing sites that define each binding pocket. In the following, pocket P01 or 1 is defined by four sites with LGFE values for each site shown in the B-factor column. The algorithm does not number pockets consecutively: in this example, there is no P02 pocket, and pocket P03 contains 2 sites.
ATOM 1 X P01 A 1 20.980 7.230 -12.513 1.00 -4.39 C ATOM 2 X P01 A 1 27.947 16.848 -6.986 1.00 -2.82 C ATOM 3 X P01 A 1 17.606 12.224 -10.043 1.00 -2.67 C ATOM 4 X P01 A 1 14.912 17.557 -5.282 1.00 -2.08 C ATOM 5 X P03 A 3 16.182 -10.455 11.793 1.00 -2.96 C ATOM 6 X P03 A 3 17.605 0.058 15.384 1.00 -2.83 C
pdb_by_pocket: Directory containing PDB files for fragments that comprise each pocket. For example,pocket_all_8_site_12_10_1.pdbindicates pocket 8 contains a fragment from site 12 and that fragment is fragment 10.1indicates that it is the first copy of fragment 10 in that pocket. REMARK includes the LGFE scores, the B-factor column includes the GFE score for each atom, and the final column is the SILCS atom type.
Note
If you encounter an error in steps 2 or 3, it is likely that required Python 3 packages have not been installed. Please refer to Python 3 Requirement for more information.
Practical considerations¶
1_setup_silcs_hotspots
creates a subdirectory, 4_hotspots/, (or user defined name using hotspotsdir=) that contains fragment pose and
LGFE information. As SILCS-Hotspots makes use of
large numbers of SILCS-MC calculations on each fragment, this directory
will be filled with a substantial amount of data. It is suggested that, once
all analyses are complete, these files either be deleted or archived,
and 4_hotspots/ be renamed prior to additional SILCS-Hotspots
runs. Remember, the data in 4_hotspots/ are used for
post-run clustering and site determination and report generation.
If 1_setup_silcs_hotspots is being rerun with new
parameters, such as new fragments as specified by ligdir=, the subdirectory
4_hotspots/ should be renamed or an alternate name assigend
using hotspotsdir= to avoid information from the original
run being overwritten. However, this is not strictly necessary IF all
the new fragments have unique filenames relative to the original run AND
the SILCS-MC job parameters specified by paramsfile= are not changed,
since the subsequent 2_collect_hotspots command only performs analysis on
Mol2/SDF files in the specified ligdir directory.
1_setup_silcs_hotspots launches a large number of jobs; while
the majority of these jobs finish quickly, individual jobs may take time
(minutes to an hour) to complete.
In some cases one or two jobs in the set may require additional time due
to the robust SILCS-MC convergence criteria used in SILCS-Hotspots.
For the SILCS-MC sampling, especially with larger fragments or ligands,
you may prefer not
to include the SILCS Exclusion Map. This will permit fragment sampling of
poses that would otherwise be rejected because of overlap with the
exclusion region. Using this approach, final fragment
poses will be based solely on scoring with SILCS FragMaps. To achieve
this, set the weighting of the Exclusion Map to zero in your custom
SILCS-MC job parameters file (paramsfile=<custom params file>).
A simple means to this end is to use a copy of
the default file $SILCSBIODIR/silcs-mc/params_custom.tmpl in which you
replace the 1.000 in the below line with 0.000.
SILCSMAP EXCL <MAPDIR>/<prot>.excl.map 1.000
Clustering of data in the 4_hotspots/ or equivalent subdirectory with the
2_collect_hotspots command produces representative fragment poses on a
per-cluster basis. If clustering is repeated, for example with a different
clustering radius, the old PDB files created in 4_hotspots/ will be
overwritten. Therefore, make sure to run site determination and report
generation (the 3_create_report command) on your original
clustering output before repeating clustering.
2_collect_hotspots processes each of the fragments individually and
typically requires minutes to complete for 100 fragments. During that process a
number of warning messages, such as “Clustered PDB not found:
4_hotspots/2/subspace_1/pdb/2_clust_1_1.pdb,” will be given. These are expected
as they indicate subspaces for the fragments in which no favorable
fragments poses
were identified. For example, this may occur where the subspace encompasses
the protein structure.
Site determination and report generation with the 3_create_report command
creates a subdirectory,
4_hotspots/report/, and outputs report.xls and PDB files into this
subdirectory. Rerunning 3_create_report with different parameters,
like an alternate value of the site clustering cutoff, will overwrite the
original output. Therefore, prior to rerunning report generation, rename
4_hotspots/report/ to save your original report generation outputs.
The clustering alogrithm for site determination does not consider the
identity of the fragment when performing the clustering. Accordingly, in
certain cases it is possible for the same fragment to be included two or
more times in a given site.
Example¶
The following demonstrates use of SILCS-Hotspots on p38 MAP kinase.
Input files, including FragMaps, are in $SILCSBIODIR/examples/silcs/.
cp -r $SILCSBIODIR/examples/silcs/silcs_fragmaps_p38a .
cd silcs_fragmaps_p38a
$SILCSBIODIR/silcs-hotspots/1_setup_silcs_hotspots prot=p38a.pdb ligdir=$SILCSBIODIR/data/databases/ring_subset mapsdir=maps bundle=true
In this example, we use $SILCSBIODIR/data/databases/ring_subset as the
fragment database (see Default databases of fragment-like molecules). Owing to the small size of
this database, the example run finishes quickly.
Once the jobs spawned by 1_setup_silcs_hotspots complete, use the
following commands for post-run clustering and site determination and
report generation.
$SILCSBIODIR/silcs-hotspots/2_collect_hotspots prot=p38a.pdb ligdir=$SILCSBIODIR/data/databases/ring_subset
$SILCSBIODIR/silcs-hotspots/3_create_report ligdir=$SILCSBIODIR/data/databases/ring_subset
This will have created the 4_hotspots/report/ subdirectory containing the
following:
hotspots_sites.pdb: Centroid positions of fragment clusters, that is, the “hotspots.” Clusters are ranked using average LGFE scores, with the cluster rank listed in the residue number field and the average LGFE score in the B-factor field.report_all.xlsx: Report of hotspots in spreadsheet format.pdbs_by_sites: List of PDB files for each hotspot.
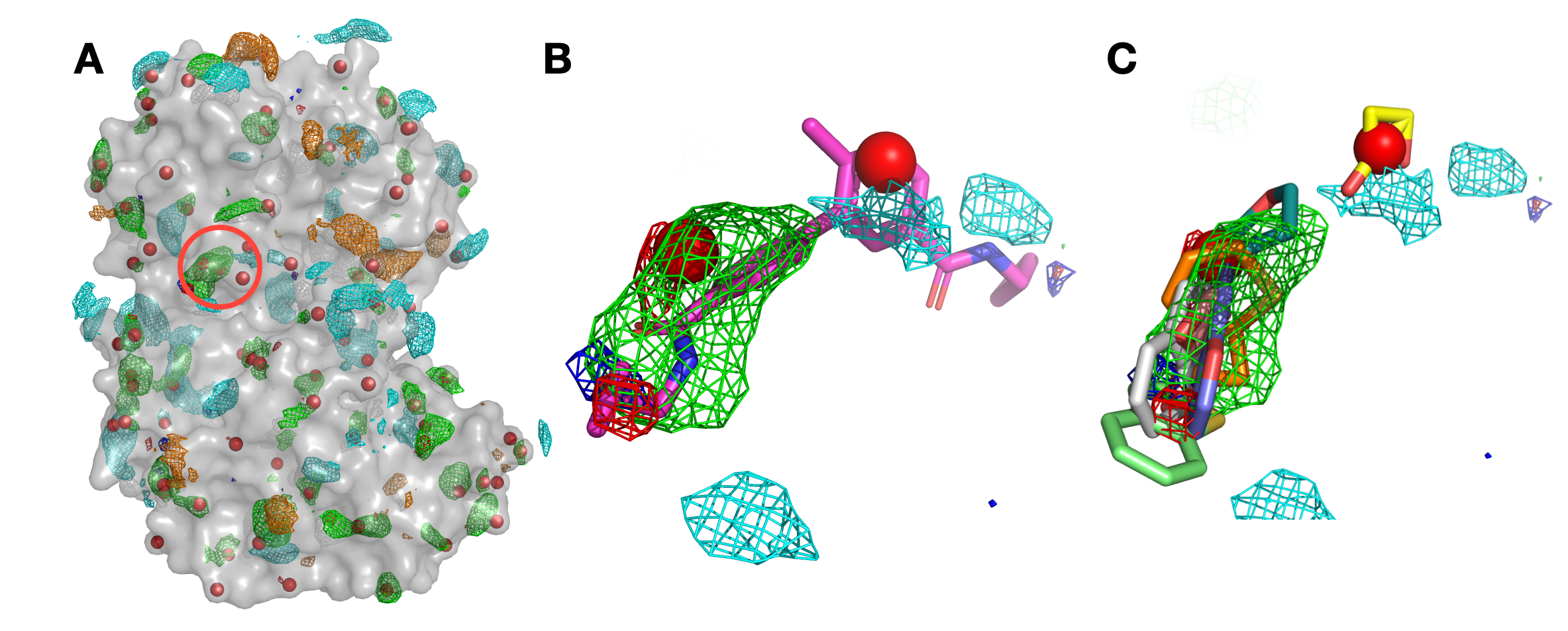
Panel (A) shows p38 (surface representation), SILCS FragMaps (wire frame), and all hotspots (red spheres) determined by SILCS-Hotspots. The crystal binding pocket is circled in red. Panel (B) has FragMaps, hotspots, and the crystallographic ligand. Two hotspots are identified within the crystal binding pocket and coincide with the rings of the ligand. Panel (C) shows fragment poses from SILCS-Hotspots calculations as well as FragMaps and hotspots.
Validating hotspots using FDA-approved drugs¶
A SILCS-Hotspots run will typically use a database of fragment-like molecules (see Default databases of fragment-like molecules) to determine probable small-molecule binding hotspots on the input target. Further validation of a subset of these hotspots can be done by docking and scoring actual FDA-approved drugs at these hotspot locations. To this end, an automated workflow is provided for docking 348 diverse FDA-approved drugs at user-selected hotspots and calculating the drug molecules’ relative buried surface areas (rBSA) before and after docking.
Note
The percent relative buried surface area (%rBSA) of a ligand is defined as
where \(\textrm{SASA}_{\textrm{unbound}}\) is the surface accessible surface area of the unbound ligand and \(\textrm{SASA}_{\textrm{bound}}\) is the surface accessible surface area of the bound ligand.
A high %rBSA of a ligand indicates that the ligand is well encased in the protein binding pocket. A low %rBSA of a ligand indicates that the ligand is bound on the surface of the protein. In general, a high %rBSA is desireable.
Suggested characteristics of hotspots to be selected for further validation¶
Users will need to judiciously choose a subset of the hotspots determined by SILCS-Hotspots for further validation by docking of FDA-appoved drugs. As these drug molecules are significantly larger and have more rotatable bonds than the fragment-like molecules used in determining hotspots, there is relatively more computational expense associated with docking them versus fragment-like molecules. This can make it impractical to apply the validation procedure to the entire set of hotspots. Suggested characteristics for choosing a hotspot to include in your subset for further validation include
The hotspot is partially or fully buried
The hotspot has other hotspots nearby (typically within 10 Å)
The hotspot has 2 or more apolar FragMap regions in close vicinity
The hotspot has multiple hydrogen bond Don/Acc FragMap regions in close vicinity
The following steps describe the SILCS-Hotspots validation procedure.
Set up and run SILCS-MC simulations at selected SILCS-Hotspots:
Once you have selected your subset of hotspots, you will need to EITHER supply them as a separate pdb file
<my_hotspots.pdb>OR as a comma-separated list (e.g."2,7,10,22") along with the fullhotspots_sites.pdbfile from the SILCS-Hotspots standard report:$SILCSBIODIR/silcs-hotspots/4a_fda_top20_analysis prot=<prot PDB> hotspotspdb=<my_hotspots.pdb>
OR
$SILCSBIODIR/silcs-hotspots/4a_fda_top20_analysis prot=<prot PDB> hotspotspdb=hotspots_sites.pdb myhotspots="2,7,10,22"
This first step will set up and run SILCS-MC simulations of each molecule in a database of 348 FDA-approved drug molecules for each selected hotspot. The SILCS-MC in this step entails exhaustive sampling of ligand orientations and conformations.
Optional parameters:
A tag identifying the subset of hotspots you are analyzing:
sitetype=<tag_name>
The default
<tag_name>is set to"rings". If you use this option, make sure to use it for ALL the following steps as well.FragMap directory path:
mapsdir=<location and name of directory containing FragMaps; default=maps>
By default, the program looks for FragMaps in the
maps/directory.Compound library/database location:
sdfile=<location and name of SDF containing all compounds; default=$SILCSBIODIR/data/databases/fda_approved_348.sdf>
or
ligdir=<location and name of directory containing compound mol2/sdf>
The default database is a database of 348 diverse FDA-approved drug molecules, which is expected to capture the chemical space of existing drug molecules. However, you may choose to supply a different database.
Option to bundle jobs:
bundle=<true|false; default=true>
Multiple (single) jobs will be bundled into a single larger job by default (
bundle=true). The number of jobs to be bundled can be set with thenprocparameter.Number of jobs to bundle:
nproc=<number of jobs to bundle; default=8>
Collect SILCS-MC results:
The second step involves collecting poses from the completed SILCS-MC simulations of the 348 FDA-approved drug molecules and rank ordering them. The collected results are output to
lgfe.csv.$SILCSBIODIR/silcs-hotspots/4b_fda_top20_analysis prot=<prot PDB> hotspotspdb=<my_hotspots.pdb>
Compute rBSA:
The third step will set up and submit jobs to compute rBSA values for top compounds.
$SILCSBIODIR/silcs-hotspots/4c_fda_top20_analysis prot=<prot PDB> hotspotspdb=<my_hotspots.pdb>
Optional parameter:
Number of top compounds for subsequent analysis
top=<number of top results for analysis; default=20>
The compounds are rank-ordered by their LGFE score in ascending order (most favorable to least favorable), and the top compounds are selected for subsequent analysis. By default, this is set to
top=20as suggested by the names of the job scripts in this section. Thus, the top 20 compounds will be selected by default. If you change this default, make sure to include the same option in the next final step.Collect final rBSA and LGFE results:
The fourth and final step will collect rBSA results, plot rBSA-vs-LGFE, and create a directory
fda_top20_analysiscontaining the final results.$SILCSBIODIR/silcs-hotspots/4d_fda_top20_analysis prot=<prot PDB> hotspotspdb=<my_hotspots.pdb>
To evaluate a different subset of hotspots, run the above steps from the begining with the new subset of hotspots and a different
sitetypevalue specified in the first step, Set up and run SILCS-MC simulations at selected SILCS-Hotspots.Warning
Re-using the same
sitetypevalue will overwrite any previous results.
Default databases of fragment-like molecules¶
Three default databases of fragment-like molecules are provided with your SilcsBio software. You may choose to use one of these for your SILCS-Hotspots run, or use your own custom molecule or database of molecules. The databases are provided both as directories containing one molecule per Mol2 file and as a single SDF file containing all molecules in the database.
$SILCSBIODIR/data/databases/{ring_system,ring_system.sdf}: Mono- and bicyclic rings commonly appearing in drug-like compounds [22].
$SILCSBIODIR/data/databases/{ring_subset,ring_subset.sdf}: A small subset of thering_systemdatabase.
$SILCSBIODIR/data/databases/{astex_mini_frag,astex_mini_frag.sdf}: Polar fragments curated from Astex [21].