SILCS-MC: Ligand Optimization¶
Background¶
The power of SILCS lies in the ability to use FragMaps to rapidly evaluate binding of diverse ligands to a target. SILCS-MC is Monte-Carlo (MC) sampling of ligands in translational, rotational, and torsional space in the field of FragMaps. MC sampling uses CGenFF force field intramolecular energies and the Ligand Grid Free Energy (LGFE), which is the sum of atomic Grid Free Energies (GFEs). The Exclusion Map prevents ligand sampling where no probe or water molecules visited during SILCS simulations. SILCS-MC allows for rapid conformational sampling of the ligand while accounting for protein flexibility in a mean-field-like fashion since ligand affinity and volume exclusion are embedded in the combination of FragMaps and the Exclusion Map. Final ligand scoring is based on the LGFE score [9][23].
FragMaps previously generated for a target (see SILCS Simulations) can be used for optimization of a parent ligand by rapidly evaluating LGFE scores for derivatives of the parent ligand. The SILCS-MC approach has two principal advantages over free energy perturbation (FEP): functional group modifications to the parent ligand are very quick to evaluate, and there is no upper size limit on these modifications. Because the calculations are done in the context of pre-computed FragMaps, it is easy to decide which functional groups are candidates for modification simply by viewing the binding pose of the parent along with the FragMaps. FragMap densities adjacent to but unoccupied by parent ligand atoms offer opportunities for growing the parent ligand. Conversely, parent ligand atoms not in favorable regions of the FragMap densities can be candidates for modification or deletion without affecting binding affinity. Quantitative evaluation of individual functional group contributions to binding affinity can be achieved by analysis of atomic GFE scores. These capabilities are particularly useful when modifying functional groups to optimize pharmacokinetic properties or attempting to reduce the ligand size while maintaining affinity.
SILCS-MC Ligand Optimization Using the SilcsBio GUI¶
Begin a new SILCS-MC Optimization project:
Select New SILCS-MC Optimization project from the Home page.
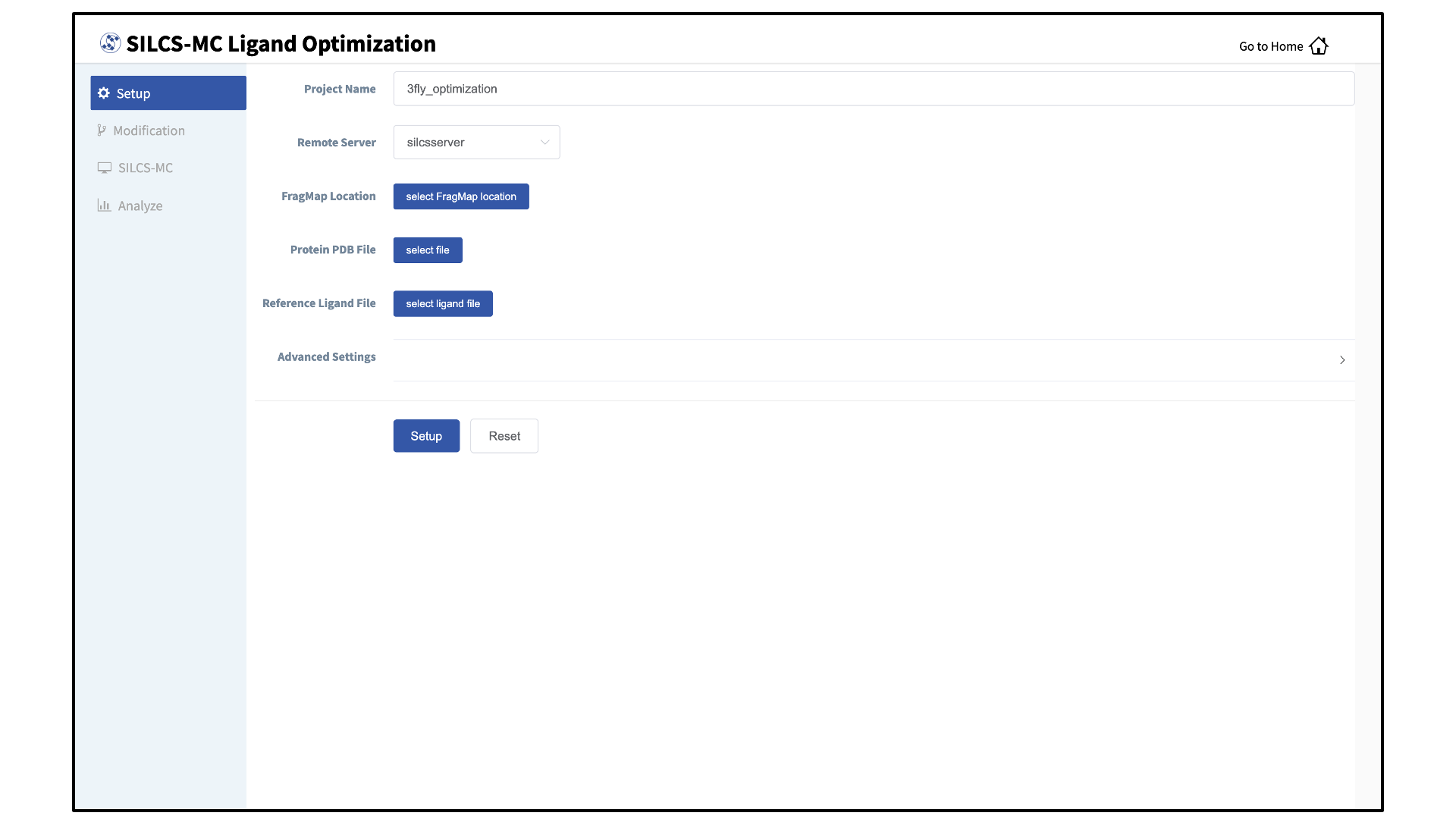
Enter a project name, select the remote server, and input files:
Enter a project name and select the remote server where your SILCS-MC jobs will run. Next, provide FragMap, protein, and ligand input files. You may choose these files from the computer where you are running the SilcsBio GUI (“localhost”) or from any server you have previously configured, as described in File and Directory Selection.
Tip
The “Reference Ligand File” must be an SD or Mol2 format file that contains the parent ligand aligned in the binding pocket of the input protein.
Upload input files to the remote server:
Once all information is entered correctly, press the “Setup” button at the bottom of the page. The GUI will contact the remote server and upload the your input files to the “Project Location” directory on the remote server. A green “Setup Successful” button will appear once the upload has successfully completed. Press this button to proceed.
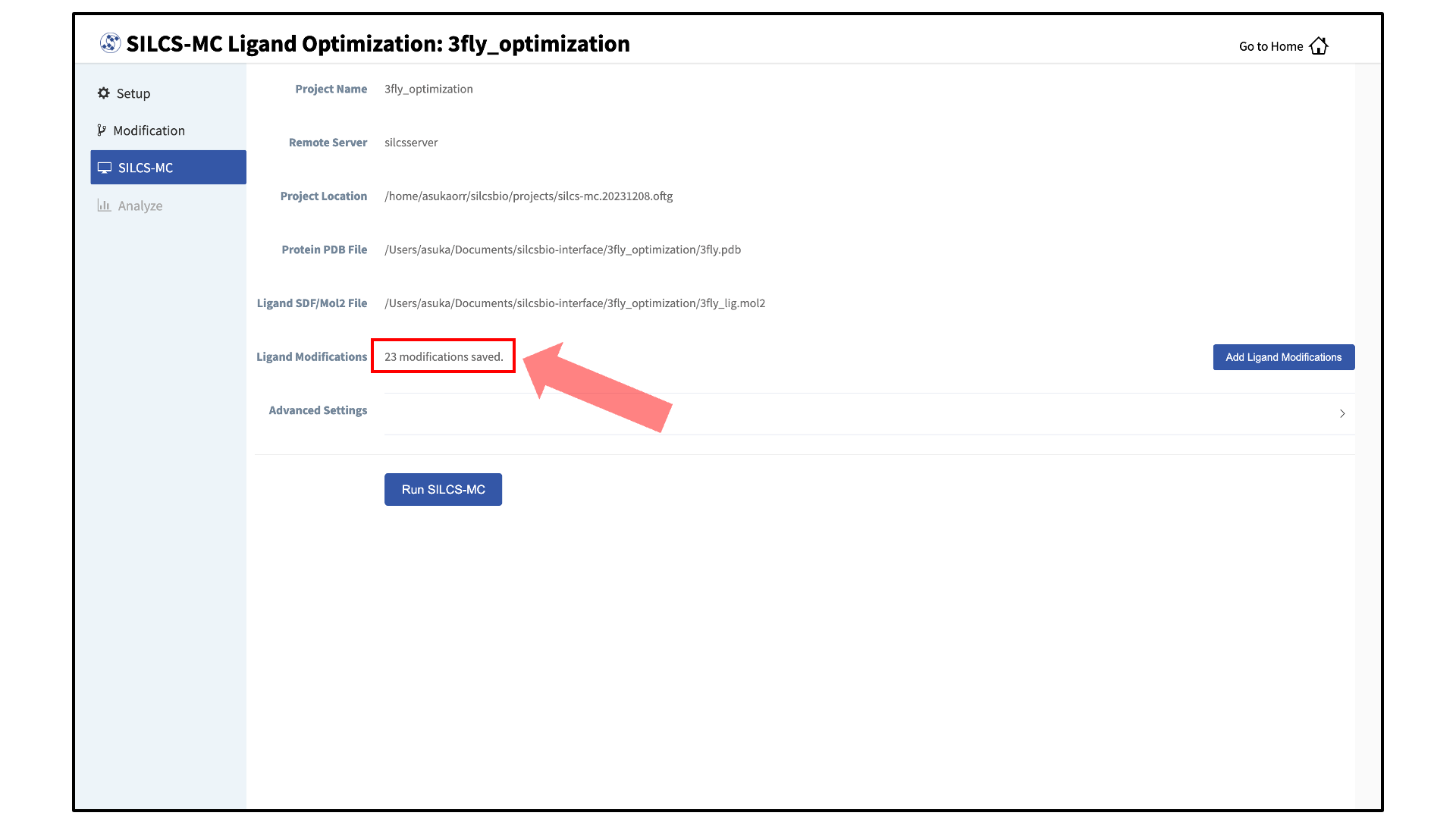
Add ligand modifications:
The GUI will display a summary screen with the Project Name, Remote Server, Project Location, Protein PDB File, Ligand SDF/Mol2 File, and Ligand Modifications. The entry for Ligand Modifications will state “0 modifications saved” and have a button labeled “Add Ligand Modifications.” Press this button to select modifications to the parent ligand for evaluation by SILCS-MC.
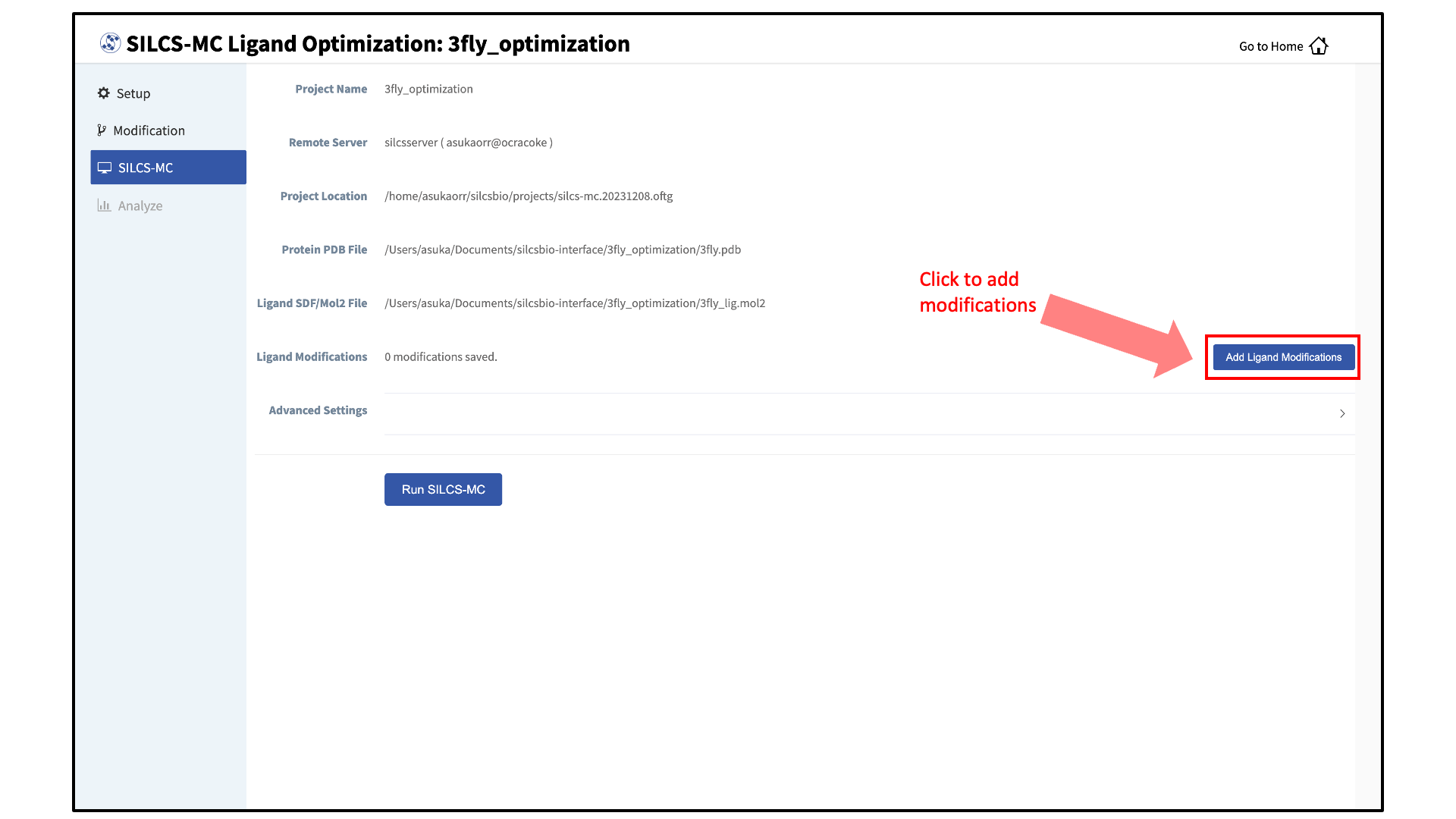
You will now see the parent ligand in the binding pocket. Rotate the view until you can easily see the functional group you wish to modify. There are two major modification types, Substitution and Replacement, available in the GUI. Substitution is used to substitute an atom with a functional group. Replacement is used to replace an atom in a ring or aliphatic acyclic group with another functional group that preserves the connectivity and valence of the ring or chain.
In the visualization window, select the atom to be modified. Then, select your desired modifications from the “Subst.” (Substitution) or the “Repl.” (Replacement) tab in the right-hand panel. Pressing the “Build Modification” button in the panel will take you to the “Review” tab.
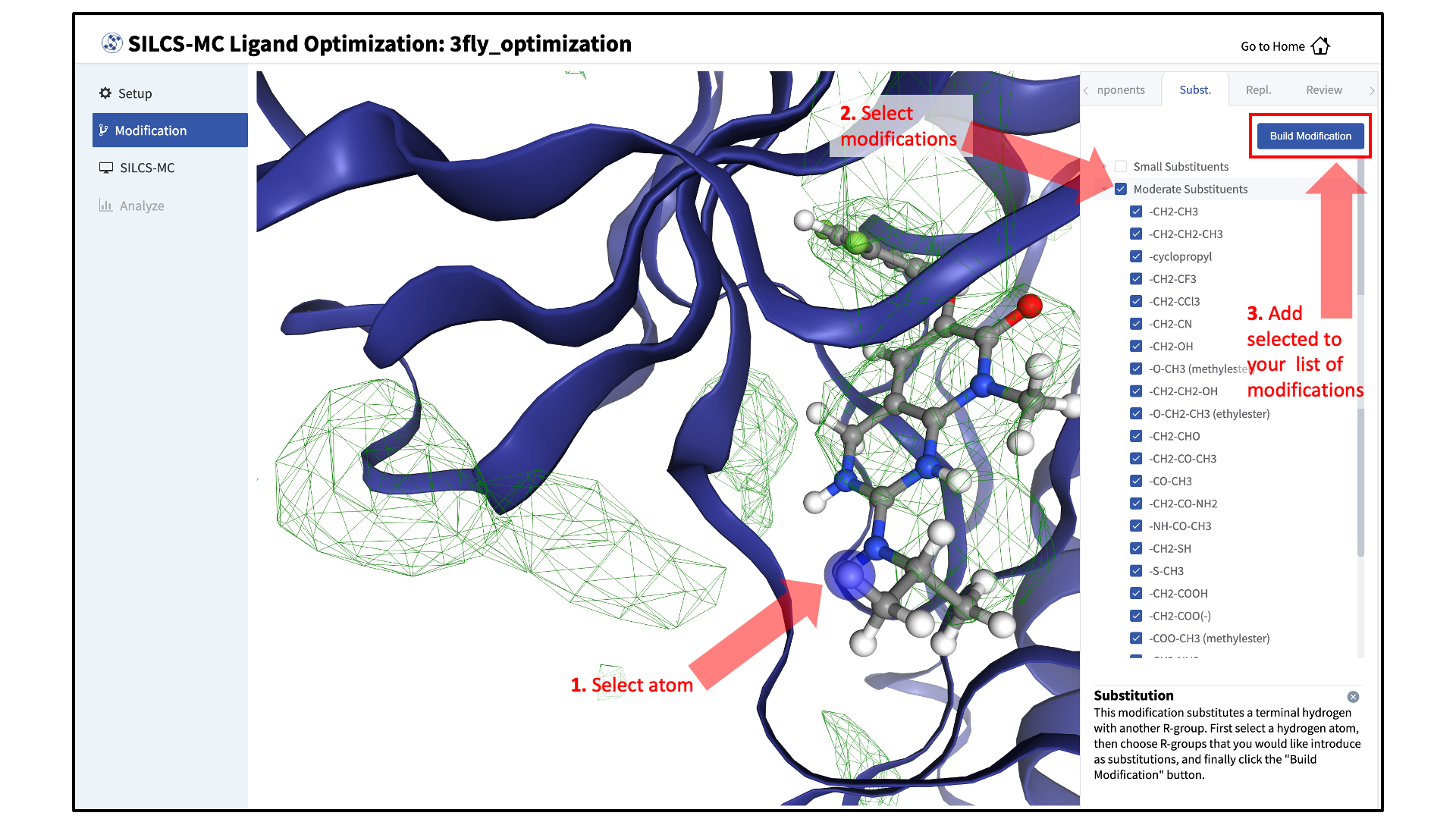
The list of modification types in the GUI covers a very broad range of chemical functionality and size.
Confirm selected ligand modifications:
Use the “Review” tab to confirm your desired modifications.
Valid modifications will have a small Image icon as well as a small Trash Can icon. Clicking on the Image icon will show the modification in the center panel. Clicking on it again will show the parent ligand. Clicking on the Trash Can icon will delete the proposed modification from your list. You can go back to the “Subst.” and “Repl.” tabs to add to your list. Once you have completed your list of modifications, you must press the “Save Modification” button in the “Review” tab to actually save the list of modifications for evaluation by SILCS-MC.
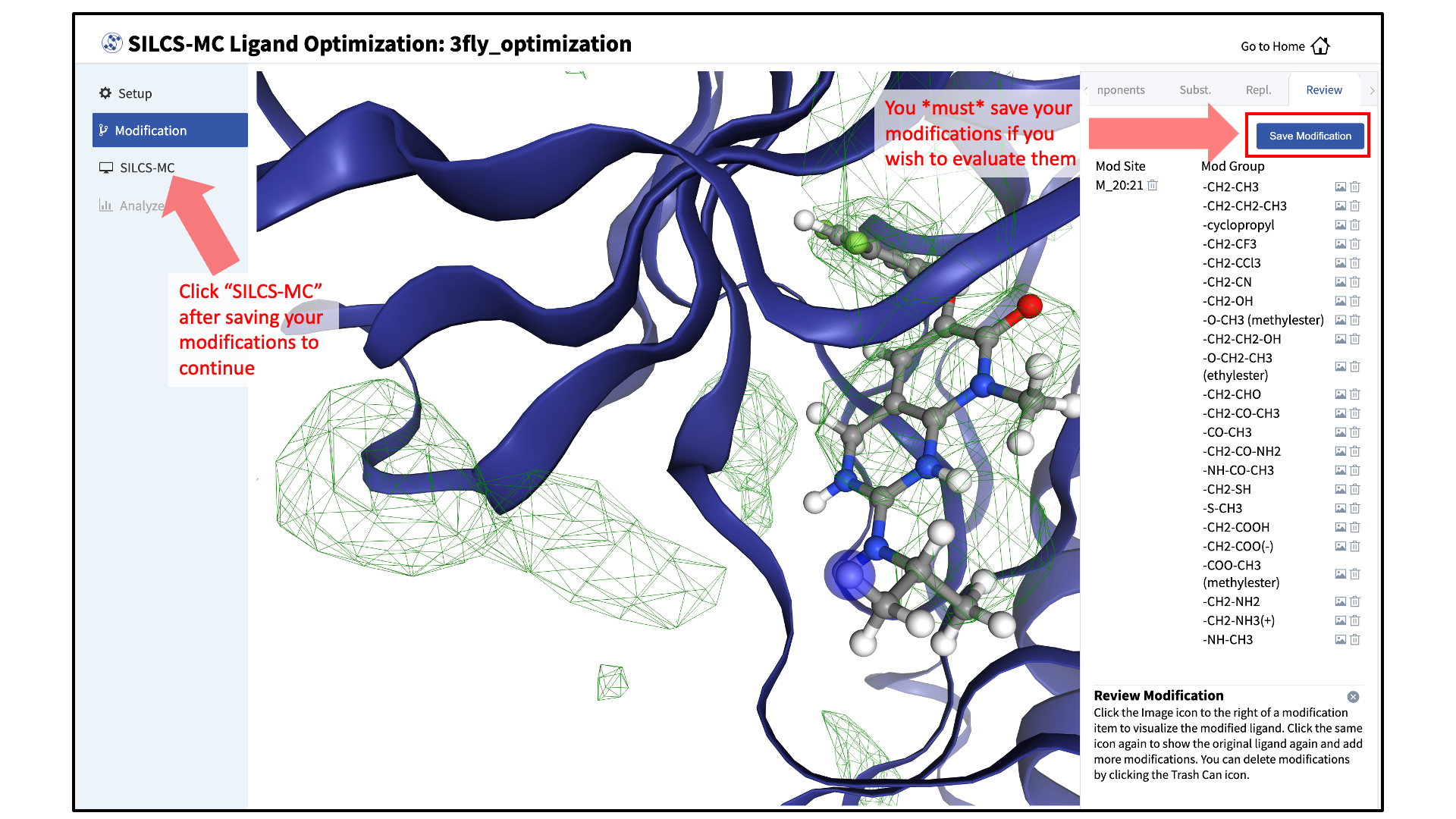
Launch SILCS-MC jobs:
Once you have saved all your desired modifications, click on SILCS-MC in the left-hand panel to go back to the summary screen. The “Ligand Modifications” will have been updated to reflect the number of saved modifications that will be evaluated by SILCS-MC. Because SILCS-MC is a fast calculation, it is feasible to select a very large number of modifications (hundreds+) for quick evaluation in a single run.
Tip
If you ran Halogen SILCS simulations for your target, you can include the Halogen SILCS FragMaps in the SILCS-MC posing and scoring by checking the “Include Halogen FragMaps” box.
Click the “Run SILCS-MC” button to launch your jobs on the remote server.
You can monitor job progress in the SilcsBio GUI, and click on the “Show Chart” button once all job progress bars are at 100%.
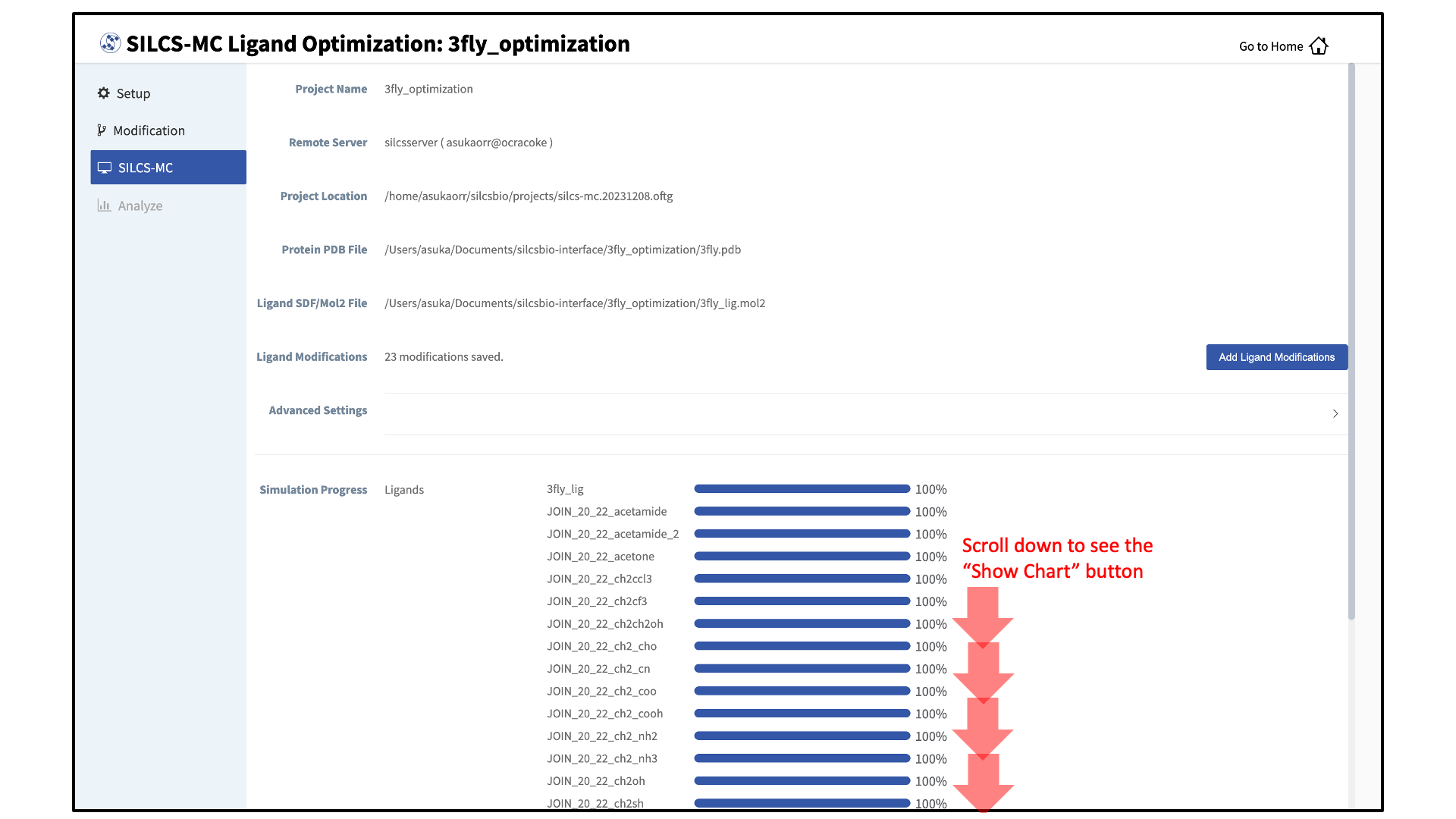
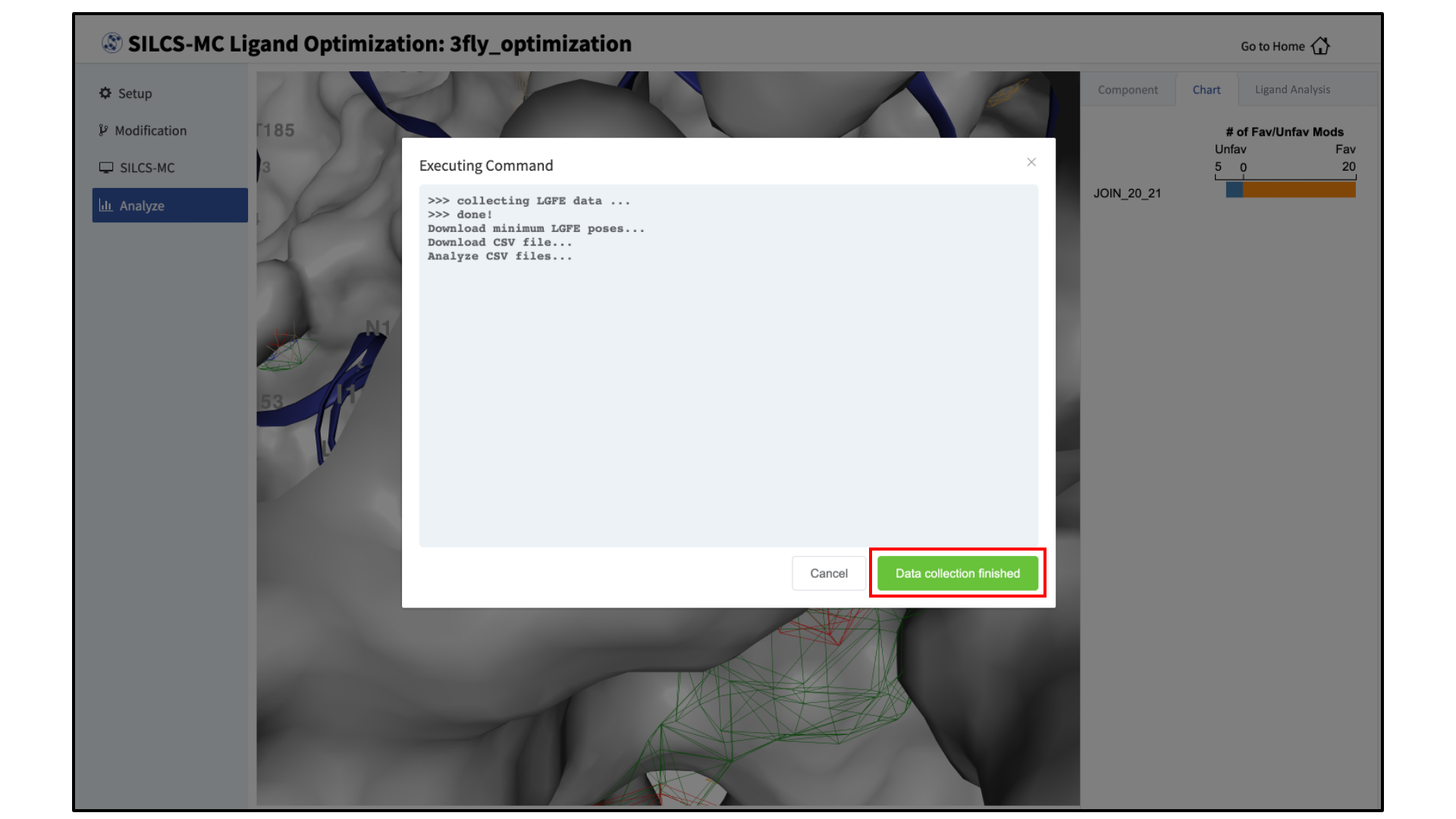
Collect SILCS-MC results:
Clicking the “Show Chart” button after jobs have reached 100% will download the SILCS-MC results from the server. Click on “Data collection finished” to proceed.
Visualize ligand modifications and change in LGFE scores:
The “Chart” tab in the right-hand panel shows a summary of the number of favorable and unfavorable modifications associated with a particular modification site. Click on the name of the modification site in that panel to see a detailed list of the change in Ligand Grid Free Energy (\(\Delta LGFE\)) relative to the parent ligand for each modification at that site.
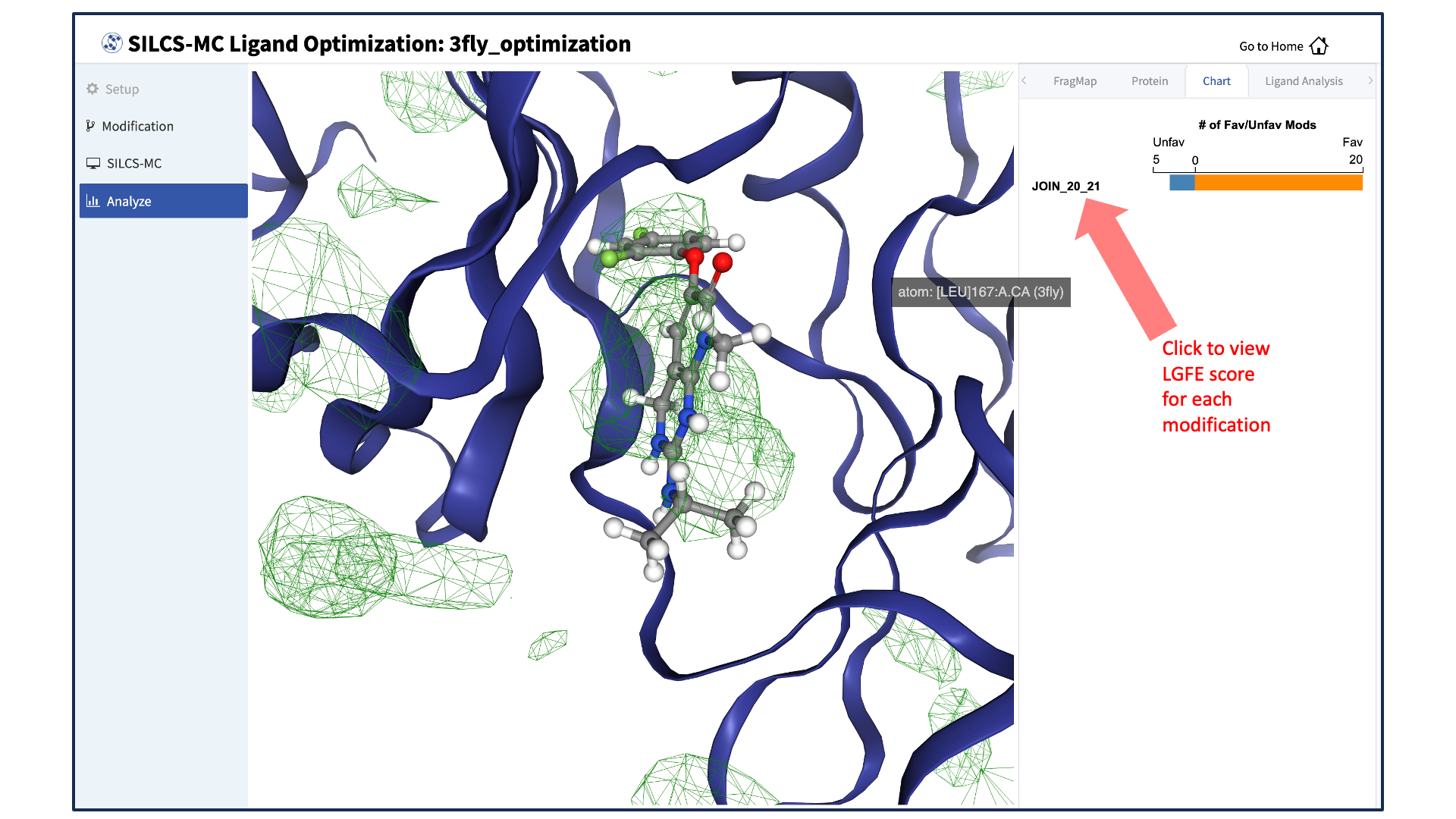
Clicking on the name of a modification will display that modification in the central panel.

The “Ligand Analysis” tab in the right-hand panel can be used to reveal the contribution of each scored atom in the ligand to the overall LGFE score. Clicking on a ligand atom in the central panel will display its atomic GFE score and add it to list of atoms that are used to compute the “Sum” of GFE values for that ligand in the right-hand panel.
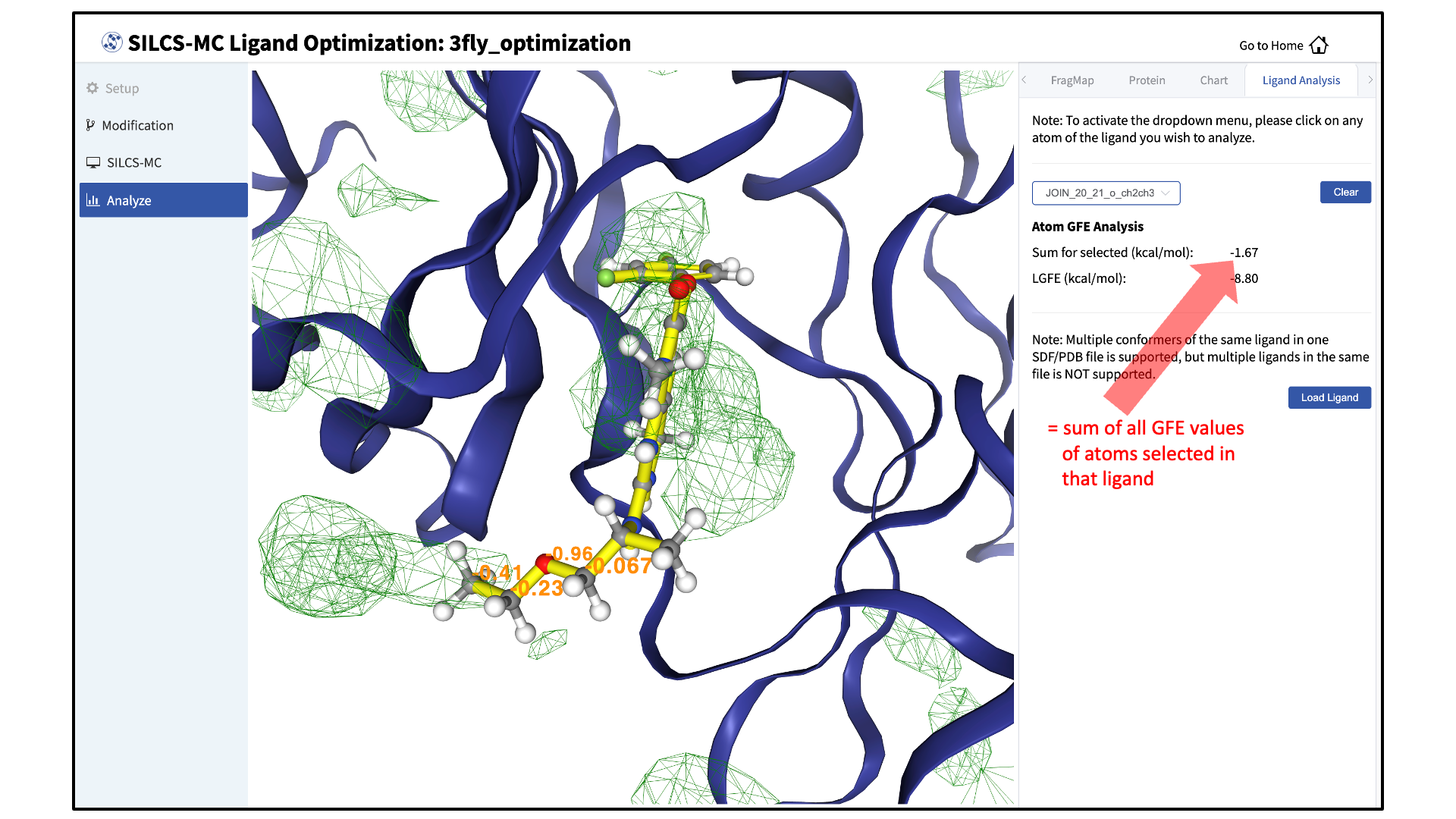
Detailed analysis of atomic GFE scores is an intuitive and powerful means to enable decisions regarding ligand modifications. Such analysis can reveal the affinity contribution of a new functional group. It can also reveal how the new functional group may have led to a change in the orientation of the ligand in the binding pocket relative to the parent compound. In such a case, comparison of atomic GFE scores for atoms preserved between the parent and derivative can reveal the preserved atoms’ contributions to the change in binding pose. See Table 2 of [24] and Figure 6 of [25] for examples.
SILCS-MC Ligand Optimization Using the CLI¶
Running SILCS-MC ligand optimization from the command line interface consists of two steps: creating a set of Mol2 files with the desired modifications to the parent ligand and then running SILCS-MC pose refinement on these files. Details for these steps can be found in reference Chemical Group Transformations and in reference SILCS-MC Pose Refinement Using the CLI, respectively.