SILCS-BML¶
Background¶
Bayesian Machine Learning (BML) Markov Chain Monte Carlo optimization allows improved predictability of the LGFE from SILCS-MC docking or pose refinement through reweighting of the FragMaps when experimental data is available for compounds in a training set. As the standard FragMaps act as priors for the optimization, the number of compounds in the training set may be relatively small (i.e., as little as 10 compounds). The ability to use a small training set allows the approach to be used in early stage ligand optimization, enabling improved predictions, and facilitates the approach to be used and updated iteratively during the drug design process, which can involve splitting the compounds into congeneric series. SILCS-BML optimization involves reweighting of the FragMap contributions to the LGFE to better reproduce selected target data. BML reweighting can target the RMSD, Pearson correlation (R), or Percent Correct (PC) metrics. The reweighting should be performed in the presence of restraints, including flat-bottom (default), hard wall, or harmonic potentials. Output from the program is directly input into a new params file to be used in subsequent SILCS-MC docking. Reweighting includes k-fold cross validation. Note that the quality of the reweighted parameters is judged based on the results from the subsequent SILCS-MC docking. In cases of overfitting, the new LGFE scores will yield poor correlations. Additional details on SILCS-BML are available in references [23] and [15]
SILCS-BML Using the CLI¶
SILCS-BML is currently available only in CLI. BML is performed in five steps as follows:
Generate fitting data:
$SILCSBIODIR/silcs-bml/1_gen_fitting_data posepdbsdir=<location and name of directory with docking poses as pdbs>The above command will set up SILCS-BML runs by generating training and validation sets based on PDB files of docked ligands under investigation and a user-provided list of experimental binding affinity data for the same ligands. The docked ligands should correspond to those generated from SILCS-MC docking, located in the
minconfpdbdirectory with GFE scores of the different classified atoms included in each PDB file.Note
SILCS-BML requires pdbs generated from SILCS-MC docking and experimental data provided as a list of experimental binding affinity data (
exp.datfile in the directory in which the1_gen_fitting_datacommand is being executed or filepath specified by the user as an optional parameter). The SILCS-MC docked poses in PDB format can be generated using thesilcs-mc/2_cals_lgfe_min_avg_sdcommand with optionpdb=true.Required parameter:
Path and name of directory containing ligand docked poses:
posepdbsdir=<location and name of directory with docking poses as pdbs>The ligand docked poses must be in PDB format derived from SILCS-MC docking. This directory will correspond to
3_silcsmc/minconfpdbif SILCS-MC was implemented with default parameters as described in SILCS-MC Docking Using the CLI.Optional parameters:
Path and name of data file containing experimental binding affinities in kcal/mol:
exp=<experimental data in same alphanumeric order as pose pdbs; default=exp.dat>The binding affinity data must be listed in a single column text file with one value per line. The data should be ordered such that the binding affinities of the ligands are listed in the same order as the pdb file names of the same ligands in alphanumeric order. By default, the file name will be
exp.dat.The number of sets used for cross-validation:
kfold=<k-fold validation; default=5>The supplied data sample will be split into a number of sets for cross-validation. By default,
kfold=5, and the sample data will be split into 5 sets.Note
The 0th set
set=0will always be prepared and it will use all data as training data for the SILCS-BML refinement.Path and name of output directory:
silcsbmldir=<location and name of output directory; default=6_silcsbml>Option to skip the confirmation of the order of the experimental data:
confirm=<the order of the exp data has been confirmed; true/false; default=false>When
confirm=false, the ligand pdb file names will be printed on the terminal in the order in which the experimental data is expected to be (alphanumeric order). The user will be prompted to cancel or continue the command after viewing the order of the ligands with eitheryto continue ornto cancel. If the user is confident in the order of the experimental data, this confirmation stage can be skipped withconfirm=true.
Run the refinement code:
$SILCSBIODIR/silcs-bml/2_run_refine posepdbsdir=<location and name of directory with docking poses as pdbs>The
2_run_refinecommand will execute the SILCS-BML fitting based on the setup from the previous step. The initial RMSD, Pearson correlation coefficient, and percent correct of the ligand LGFEs and experimental binding affinities will be printed on the terminal next to the final RMSD, Pearson correlation coefficient, and percent correct of the final LGFEs optimized by reweighting FragMap contributions targeting the user-defined metric (target). Additionally, the sets with the best RMSD (lowest RMSD), Pearson correlation coefficient (highest R), and percent correct (highest PC) will be printed on the terminal. This information will also be saved in6_silcsbml/2_run_refine.out.Required parameters:
Path and name of directory containing ligand docked poses:
posepdbsdir=<location and name of directory with docking poses as pdbs>The ligand docked poses must be in PDB format derived from SILCS-MC docking. This directory should correspond to the same directory specified in the first
$SILCSBIODIR/silcs-bml/1_gen_fitting_datastep.Optional parameters:
Path and name of output directory:
silcsbmldir=<location and name of output directory; default=6_silcsbml>The path and name of the output directory should correspond to the same path and name specified in the first
$SILCSBIODIR/silcs-bml/1_gen_fitting_datastep.Target metric for optimization (integer):
target=<choose optimization target (1: RMSD | 2: Pearson Correlation | 3: Percent correct); default=3>SILCS-BML allows user to define the target property to be optimized with 3 options to choose from: Root mean square deviation (RMSD) with
target=1, Pearson correlation (R) withtarget=2, or percent correct (PC) withtarget=3.Option to scale weights to yield original LGFE range (integer):
scale=<scale weights to yield original LGFE range (1:Yes | 0:No); default=1>The SILCS-BML fitting can be constrained such that the resulting LGFE values of the ligands are in the same range of the original set of LGFE values. This scaling can be turned off (
scale=0) or kept on (scale=1).Restraint type:
restraint=<choose restraint type (1: Flat-bottom | 2: Hard wall | 3: Harmonic); default=1>Currently, 3 types of restraint are offered: flat-bottom (
restraint=1), hard wall (restraint=2), and harmonic (restraint=3). By default, flat-bottom restraints are used (restraint=1).The restraint types are described in further detail below for weighting factor \(x\), user defined force constant \(\kappa\), and user-defined upper and lower limits of \(x\), \({x_u}\) and \({x_l}\), respectively. User-defined parameters \(\kappa\), \({x_u}\), and \({x_l}\) can be defined using the
kappa,upper, andloweroption bulletted below, after the description of restraint types.[1]. Flat-bottom potential restraint (
restraint=1)\[ \begin{align}\begin{aligned}y = \kappa (\frac{1}{1+\exp{\frac{x-x_l}{d}}}) \textrm{ if}, x < 1\\y = \kappa (\frac{1}{1+\exp{\frac{x_u-x}{d}}}) \textrm{ if}, x > 1\end{aligned}\end{align} \]Where \(d\) determines how flat the bottom is between \({x_l}\) and \({x_u}\) limits. The value of \(d\) is fixed at 0.01.
Below is a plot showing this restraint for \(\kappa = 1\) (
kappa=1), \({x_l} = 0.6\) (lower=0.6), and \({x_u} = 1.4\) (upper=1.4):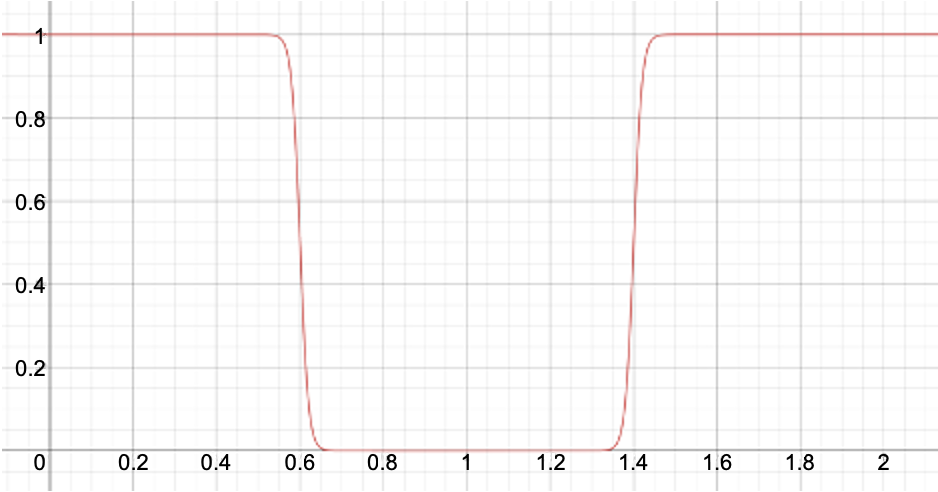
[2]. Hard-wall potential restraint (
restraint=2)\[ \begin{align}\begin{aligned}y = \mathrm{0} \textrm{ if}, \mathrm{lower < x < upper}\\y = \kappa \textrm{ else}\end{aligned}\end{align} \]Below is a plot showing this restraint for \(\kappa = 1\) (
kappa=1), \({x_l} = 0.6\) (lower=0.6), and \({x_u} = 1.4\) (upper=1.4):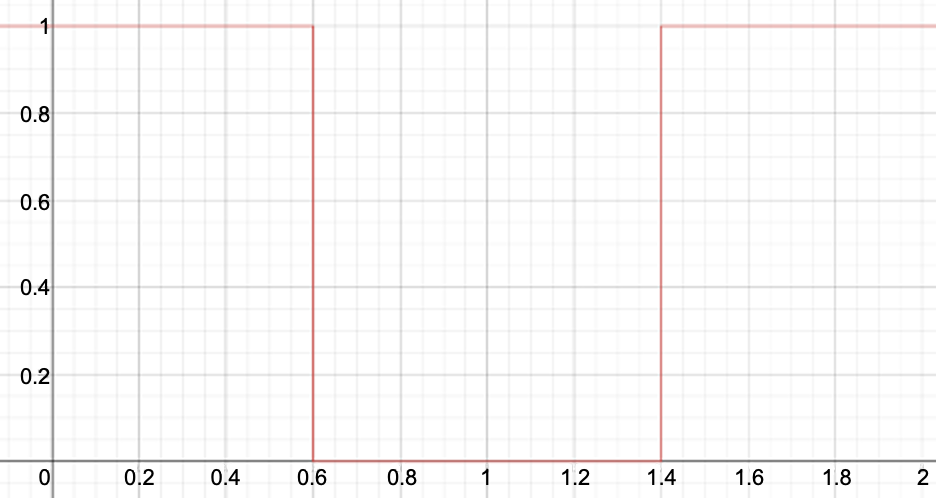
[3]. Harmonic potential restraint (
restraint=3)\[y = \kappa (x-1)^2\]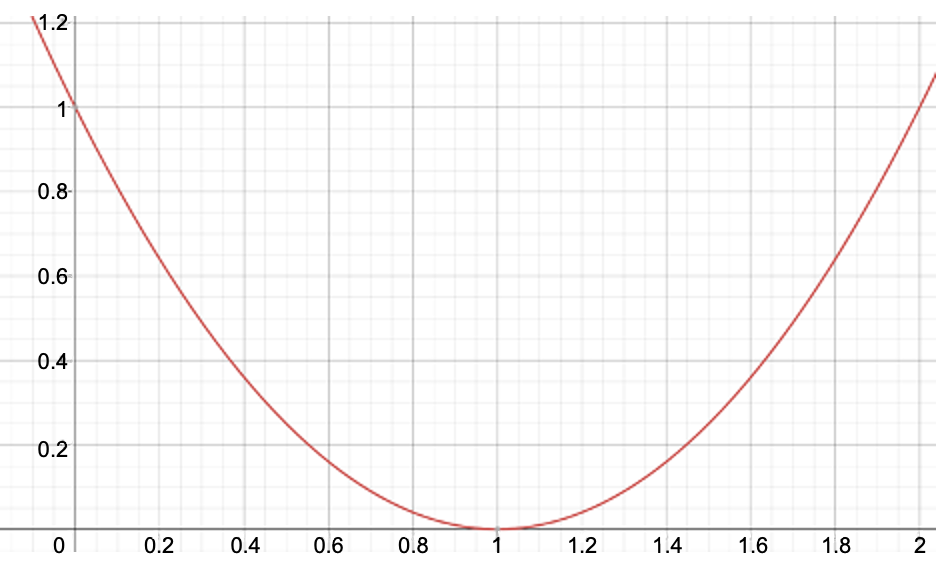
Restraint force constant (integer):
kappa=<force constant for restraint; default=300>Upper limit of weighting factor (decimal):
upper=<upper limit of weighting factor for restraint type 1 or 2; default=2>Lower limit of weighting factor (decimal):
lower=<lower limit of weighting factor for restraint type 1 or 2; default=0.1>Minimum percentage of compounds with specific FragMap type to be included in optimization (decimal):
mincmpds=<minimum percentage of compounds with specific FragMap type for that type to be included in the optimization; default=30>This option selects FragMap types to be fully optimized based on a minimum percentage of compounds with that specific FragMap type. FragMap types represented by less than the defined percentage will be optimized to a limited extent defined as (0.8~1.2) scaling of the input weight of that FragMap type. This prevents overfitting of underrepresented FragMaps. By default,
mincmpds=30.
Generate SILCS-MC Parameter File:
$SILCSBIODIR/silcs-bml/3_gen_silcsmc_inp posepdbsdir=<location and name of directory with docking poses as pdbs>After determining the optimal FragMap weighting in the second step, the ligands can be re-docked using SILCS-MC parameter files reflecting the tuned FragMap weights. The SILCS-MC parameter file with the optimal FragMap weighting for each cross-validation set (
params_reweight_*.inp) can be generated using the above command. The number ofparams_reweight_*.inpfiles will correspond to the number of cross-validation sets specified using thek-foldparameter in the first1_gen_fitting_datastep.Required parameter:
Path and name of directory containing ligand docked poses:
posepdbsdir=<location and name of directory with docking poses as pdbs>The ligand docked poses must be in PDB format derived from SILCS-MC docking. This directory should correspond to the same directory specified in the
$SILCSBIODIR/silcs-bml/1_gen_fitting_dataand$SILCSBIODIR/silcs-bml/2_run_refinesteps.Optional parameters:
Path and name of directory containing SILCS-BML fitting output:
silcsbmldir=<location and name of output directory; default=6_silcsbml>The path and name of the output directory should correspond to the same path and name specified in the previous two steps.
Path and name of SILCS-MC parameter file:
paramsfile=<silcs-mc params.inp file to be reweighted; default=params.inp>The
3_gen_silcsmc_inpcommand will edit theparams.inpfile (typically3_silcsmc/params.inpfrom original SILCS-MC docking) that was used to dock and produce the poses stored in the directory specified for theposepdbsdirparameter. The new parameter files will be stored in6_silcsbml(or the user-specifiedsilcsbmldir) asparams_reweight_*.inp.
Run SILCS-MC with the BML optimized FragMaps:
$SILCSBIODIR/silcs-bml/4_run_silcsmc_bml prot=<prot PDB> ligdir=<location and name of directory containing ligand mol2/sdf>The above command will initiate SILCS-MC docking runs using the new parameter file(s) (
params_reweight_*.inp) generated in the previous step, which specifies the reweighted FragMaps for the LGFE calculations. By default, the parameter set with the best percent correct will be used. The user can specify which parameter set to use with the optionalrunsetparameter.Required parameters:
Path and name of target protein PDB file:
prot=<prot PDB>Path and name of directory containing ligand structure files:
ligdir=<location and name of directory containing ligand mol2/sdf>Typically,
ligdircan be the same directory as theposepdbsdirparameter used in the previous1_gen_fitting_datastep assuming the user has used the standard3_silcsmc/minconfpdbdirectory as theposepdbsdirparameter.If the
ligdirparameter is used, each molecule must be stored in its own individual file (Mol2 or SDF file) as only one molecule per file in the specifiedligdirwill be processed for SILCS-MC. If the user has an SD/SDF file containing multiple molecules, use thesdfileparameter instead of theligdirparameter.Optional parameters:
Specify cross-validation set SILCS-MC parameter file (
params_reweight_*.inp) to use:runset=<set # to run silcs-mc with re-weighted paramfile; All / BestPC / BestR / No. between [0-kfold]; default=BestPC>By default, the
paramsfileoriginating from the cross-validation set with the best final percent correct will be used. The user can choose to specify which set to use with the optionalrunsetparameter. The user can choose to perform SILCS-MC docking using theparams_reweight_*.inpfile(s) generated for:
all (
runset=All) cross-validation setsonly the best percent correct (
runset=BestPC) cross-validation setonly the best Pearson correlation coefficient (
runset=BestR) cross-validation seta specific cross-validation set (
runset=<any integer number from 0 to k-fold>)Path and name of directory containing SILCS-BML fitting output:
silcsbmldir=<location and name of output directory; default=6_silcsbml>The path and name of the output directory should correspond to the same path and name specified in the previous three steps.
Path and name of directory containing FragMaps:
mapsdir=<location and name of directory containing FragMaps; default=maps>Path and name of SD file containing all ligands:
sdfile=<location and name of SD/SDF file>Option to bundle jobs:
bundle=<bundle multiple (single) jobs into larger jobs; default=true>Number of jobs to bundle into one:
nproc=<number of jobs to bundle; default=8>
Collect LGFE and poses from SILCS-MC simulations:
$SILCSBIODIR/silcs-bml/5_calc_lgfe_bml ligdir=<location and name of directory containing ligand mol2/sdf>After the SILCS-MC docking simulations are complete, the above command will collect the resulting LGFE and docked poses of all ligands. The new RMSD, Pearson correlation coefficient, and percent correct of the re-docked ligands’ LGFE scores versus the experimental binding affinities will be printed to the terminal. The following output files will be generated for the user to review:
6_silcsbml/report_bml_<runset>.csvcontaining the same information as printed on the terminal
6_silcsbml/3_silcsmc_bml_<runset>/minconfpdbcontaining the docked poses of the ligands
6_silcsbml/3_silcsmc_bml_<runset>/lgfe.csvcontaining the new ligand LGFE scores
6_silcsbml/report_bml_<runset>.pngcontaining a correlation plot of the the experimental binding affinities versus ligand LGFE scores before and after SILCS-BML fittingRequired parameter:
Path and name of directory containing ligand structure files:
ligdir=<location and name of directory containing ligand mol2/sdf>The same
ligdiras the previous step should be used unless the user has specifiedsdfileinstead ofligdirin the previous step.Optional parameters:
Specify cross-validation set SILCS-MC parameter file (
params_reweight_*.inp) to use:runset=<set # to run silcs-mc with re-weighted paramfile; All / BestPC / BestR / No. between [0-kfold]; default=BestPC>The
runsetparameter should be the same as the previous4_run_silcsmc_bmlstep. Description of therunsetparameter can be found in the previous step.Path and name of directory containint SILCS-BML fitting output:
silcsbmldir=<location and name of output directory; default=6_silcsbml>The path and name of the output directory should correspond to the same path and name specified in the previous step.
Path and name of SD file containing all ligands:
sdfile=<location and name of SD file>Number of top scoring poses to be collected:
npose=<number of best scoring poses sorted by LGFE; default=1>Option to output poses in PDB format:
pdb=<write PDB file in minconfpdb folder; true/false; default=false>When the
pdbkeywoad is set totrue, the top scoring poses will be saved in PDB format along with SDF file format. By default,pdb=false.Option to print ligand SMILES strings in output
6_silcsbml/3_silcsmc_bml_<runset>/lgfe.csvfile:smiles=<write SMILES string to lgfe.csv; default=false>If
smiles=trueis used, the6_silcsbml/3_silcsmc_bml_<runset>/lgfe.csvoutput file will contain a column with the SMILCS string of each ligand.Calculate 4DBA descriptors and LGFE-4DBA scores:
fourdba=<calc 4 descriptors and LGFE-4DBA score; true/false; default=false>The bioavailablility of compounds can be evaluated using 4D Bioavailability (4DBA) scores. Compounds with higher 4DBA scores are more likely to have oral drug-like characteristics When
fourdba=trueadditional calculations will be performed to calculate the 4DBA score for the ligands in addtion to LGFE score and LGFE-4DBA score. Please refer to 4D Bioavailability (4DBA) Calculation for additional details on 4DBA scores.Path to python executable:
python=<path to python executable>If the default path to python does not include the necessary modules, the path to the python executable with the necessary module installed must be specified with the
pythonparameter. The default python path can be checked using thewhich pythoncommand. Alternatively, entering the$SILCSBIODIR/silcs-bml/5_calc_lgfe_bmlcommand will output the default path for python. The matplotlib package is required for plotting. Iffourdba=true, then the rdkit, tqdm, ipython and scipy packages are also required. Please refer to Python 3 Requirement for more information.
Note
Note that once the new params_reweight_*.inp files with the optimized FragMap
weighting factors for each cross-validation set is produced it may be used for
SILCS-MC docking and pose refinement using the 1_run_silcsmc_custom command and
specifying paramsfile=params_reweight_<best performing cross-validation set>.inp
where params_reweight_*.inp is from step 3 (3_gen_silcsmc_inp) above.